
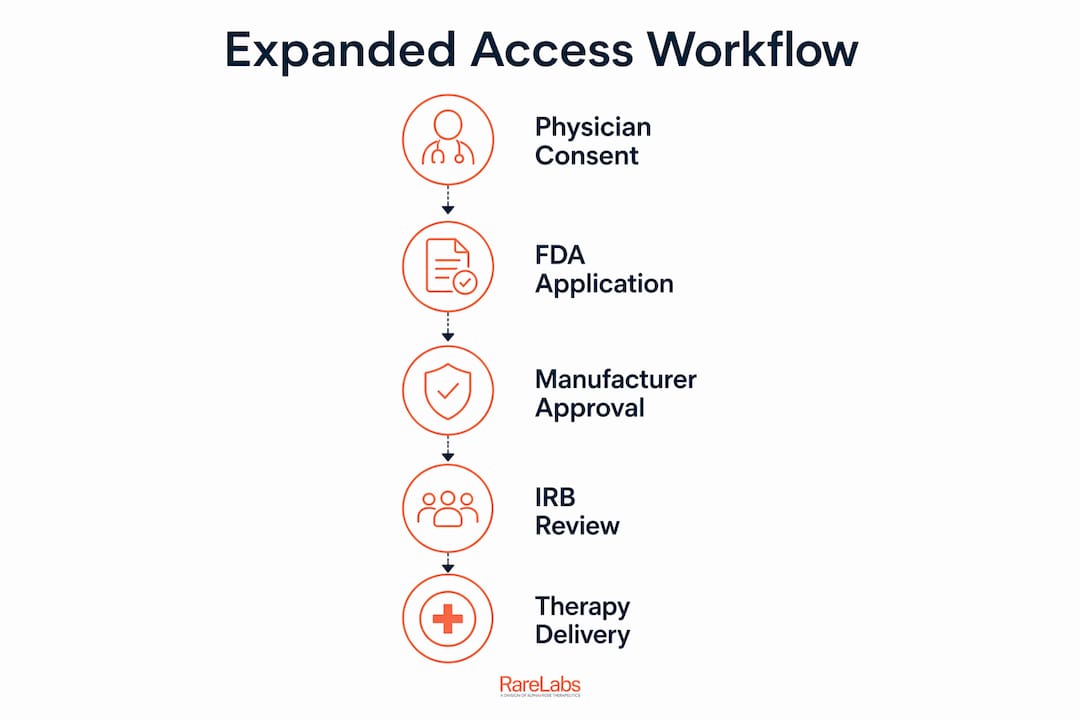
The workflow for accessing experimental therapies is a defined, multi-step process that requires physician authorization, FDA expanded access submission, manufacturer agreement, and Institutional Review Board (IRB) approval. For patients with rare genetic diseases, this process goes by several formal names: "expanded access," "compassionate use," and "individual patient IND." All refer to the same regulatory pathway that allows patients outside of clinical trials to receive investigational drugs. The FDA, treating physicians, and drug manufacturers each play a distinct, non-negotiable role. Understanding who does what, and in what order, is the difference between months of confusion and a clear path forward.
What is the workflow for accessing experimental therapies?
The pathway to new therapies outside of approved treatments runs through the FDA's expanded access program. This program allows patients with serious or life-threatening conditions to receive investigational drugs when no satisfactory approved alternatives exist. The FDA approves over 99% of individual patient expanded access requests that are complete and compliant. That statistic means the bottleneck is rarely the FDA itself. The real obstacles are preparation, physician engagement, and manufacturer cooperation.
Three entities must align before a patient receives an investigational drug. The treating physician files the application. The FDA reviews it. The drug manufacturer agrees to supply it. Each step depends on the one before it. Patients and families who understand this sequence avoid the most common delays.

What prerequisites do you need before starting the process?
Preparation is the stage most families underestimate. Getting it right shortens the entire timeline by weeks.
Patient eligibility is the starting point. The FDA requires that the patient has a serious or life-threatening condition and that no satisfactory approved therapy exists. For most rare genetic disease patients, this threshold is met. Gathering documentation to prove it is the practical task.
Required preparation steps:
- Compile complete medical records, including genetic test results, biopsy reports, and prior treatment history.
- Obtain a confirmed diagnosis from a specialist, ideally with supporting molecular or genomic data.
- Identify a treating physician who is willing to file an Individual Patient IND using FDA Form 3926.
- Clarify insurance coverage early. Expanded access is distinct from clinical trials, and insurance coverage is often limited, making significant out-of-pocket costs probable.
- Research the specific drug or therapy you are requesting, including its current development stage and the manufacturer's contact information.
The physician role is the most critical prerequisite. Treating physicians act as gatekeepers and must commit significant time and accept legal liability for filing and supervising experimental treatment. Not every physician is familiar with this process. Finding one who is, or preparing materials that make the process easier for your current physician, is a task families must take on themselves.
Pro Tip: Prepare a one-page patient summary that includes diagnosis, prior treatments, genetic findings, and a brief rationale for the specific experimental therapy. Physicians who receive organized packets are far more likely to engage with the IND filing process.
Only about 5% of eligible patients initiate discussions about clinical trials or experimental options, often because they assume their physician will raise the subject. That assumption costs time. You must ask directly.
How does the step-by-step expanded access filing process work?
The process follows a fixed sequence. Skipping or reordering steps causes rejections and delays.
- Physician completes FDA Form 3926. This is the Individual Patient IND application. It requires a clinical rationale, a proposed treatment plan, risk assessment, and informed consent documentation.
- Simultaneous IRB submission. The physician submits the single-patient protocol to an Institutional Review Board for ethics review at the same time as the FDA submission. Synchronizing FDA IND submission and IRB approval significantly reduces delays.
- FDA review period. The standard non-emergency review takes 30 days. During this window, the FDA may request additional information or clarification.
- Manufacturer agreement. While FDA review is underway, the physician or patient advocate contacts the drug manufacturer to request supply. This step runs in parallel, not after FDA approval.
- IRB approval confirmation. The IRB issues its approval for the single-patient protocol, which is required before treatment can begin.
- Drug shipment and treatment initiation. Once all three approvals are in place, the manufacturer ships the drug and treatment begins under physician supervision.
- Ongoing monitoring and reporting. The physician monitors the patient and submits required safety reports to the FDA throughout the treatment period.
Emergency situations follow a faster track. The FDA can authorize emergency use of an investigational drug via telephone or electronic communication, bypassing the standard 30-day review entirely. Written documentation must then be submitted to the IRB within 5 working days of use. This pathway exists for patients who cannot wait a month for standard review.
The end-to-end process for personalized experimental therapies typically takes about 4–5 months from initial biopsy to first treatment dose. That timeline includes clinical evaluation, sequencing, manufacturer agreement, FDA and IRB submission, and drug shipment. Planning for this duration from the start prevents families from being blindsided midway through.
What are the most common delays and how do you avoid them?
Knowing where the process breaks down is as useful as knowing the steps themselves.
Manufacturer refusal is the single most unpredictable obstacle. Drug manufacturers decide voluntarily whether to provide investigational drugs, and the FDA cannot compel them to do so. A clear, specific rationale for why this patient is a strong candidate improves the chances of agreement. Generic requests rarely succeed. Patient-specific data, including genetic findings and failed prior treatments, makes the case concrete.
IRB unfamiliarity adds weeks to timelines. Regulatory delays often stem from IRB inexperience with expedited single-patient review. Selecting a hospital or academic medical center with a dedicated rare disease or oncology IRB reduces this risk substantially.
Physician reluctance is common and understandable. Filing an IND is time-consuming and carries legal responsibility. Physician reluctance often slows access, but bringing organized regulatory materials to appointments changes the dynamic. Some families work with patient advocacy organizations to prepare these packets.
Common delay triggers to watch for:
- Incomplete Form 3926 submissions, which trigger FDA requests for additional information and restart the clock.
- Contacting the manufacturer after FDA approval rather than simultaneously, adding weeks to the timeline.
- Choosing an IRB with no experience in single-patient protocols.
- Confusing expanded access eligibility with clinical trial eligibility, which uses different criteria.
Pro Tip: Contact the drug manufacturer's medical affairs department before your physician files the IND. Confirming supply availability in advance prevents the worst-case scenario: full regulatory approval with no drug to administer.
The steps for therapy access in rare disease cases reward families who treat the process like a project. Track every submission date, follow up on every pending item, and document every conversation with manufacturers and IRBs.
How do you find and evaluate experimental therapies for rare genetic diseases?
Finding the right therapy to request is a prerequisite to filing anything. The search process has two reliable starting points.
ClinicalTrials.gov is the official U.S. registry for clinical studies. Every legitimate trial must be registered there. Trials not registered on legitimate databases should be viewed with caution. The database is comprehensive but technically dense. Resources that translate eligibility criteria into plain English improve treatment matching success and reduce the time families spend interpreting complex inclusion and exclusion criteria.
The difference between clinical trials and expanded access matters. Clinical trials collect data and follow strict protocols designed around research goals. Expanded access is focused entirely on treating the individual patient. A patient who does not qualify for a trial may still qualify for expanded access using the same drug. These are parallel pathways, not sequential ones.
| Feature | Clinical trial | Expanded access |
|---|---|---|
| Primary goal | Collect research data | Treat individual patient |
| Eligibility | Protocol-defined criteria | Serious condition, no alternatives |
| IRB oversight | Required | Required |
| FDA involvement | IND for the trial | Individual patient IND |
| Cost coverage | Often covered by sponsor | Often out-of-pocket |
When evaluating a specific therapy, check four things: the drug's current development phase, the manufacturer's expanded access policy, the geographic availability of a qualified treating physician, and the realistic logistics of monitoring and follow-up. A therapy that requires weekly infusions at a specialized center 500 miles away is a different commitment than an oral medication.
Hopeatrarelabs supports this search process directly. The platform screens thousands of FDA-approved drugs and investigational compounds against patient-specific disease models, giving families a scientifically grounded shortlist rather than a raw database to sort through. For families facing ultra-rare or undiagnosed genetic diseases, that kind of personalized research changes what is possible.
Key Takeaways
The most effective approach to accessing experimental therapies is to prepare thoroughly, engage your physician early, and run FDA, IRB, and manufacturer processes in parallel rather than sequentially.
| Point | Details |
|---|---|
| Physician engagement is first | Find a physician willing to file FDA Form 3926 before any other step begins. |
| Parallel submissions save time | Submit to the FDA and IRB simultaneously, and contact the manufacturer during review. |
| Manufacturer agreement is voluntary | Present patient-specific genetic and clinical data to improve your chances of supply approval. |
| Emergency access is faster | FDA can authorize emergency use by phone; written IRB follow-up is due within 5 working days. |
| Plan for 4–5 months | The full process from biopsy to first dose typically takes 4–5 months under normal conditions. |
What I've learned about families who actually get through this process
The families who successfully access experimental therapies share one trait: they treat the process like a second job. They do not wait for their physician to bring it up. They research the drug, contact the manufacturer, and walk into appointments with a prepared packet. That level of preparation is not optional for rare disease cases. It is the mechanism.
The regulatory framework has genuinely improved. The 2026 IRB and IND synchronization updates have reduced one of the most frustrating delays in the process. Emergency access mechanisms have also become more accessible. These are real improvements, and they matter.
What has not changed is the emotional weight of this process. Families are doing administrative and regulatory work during some of the hardest months of their lives. Advocacy organizations and platforms like Hopeatrarelabs exist precisely to reduce that burden. Using them is not a shortcut. It is the right call. The rare disease research challenges that slow access are real, but they are not insurmountable when families have the right support.
Patience and persistence are not opposites here. You need both. Push hard on every pending item, follow up weekly, and give yourself permission to ask for help from people who have done this before.
— John
How Hopeatrarelabs supports your search for experimental treatments
Hopeatrarelabs was built for exactly this situation: a patient with a rare or undiagnosed genetic disease, a family that needs answers fast, and a treatment landscape with no approved options.
The RareLabs platform creates patient-specific disease models from your own cells using iPSC and CRISPR technology, then screens thousands of FDA-approved drugs, custom antisense oligonucleotides (ASOs), and gene therapy options against that model. The result is a scientifically grounded shortlist of candidates worth pursuing through expanded access or clinical trials. Families get a rare disease treatment search tool that goes far beyond a database. The platform also offers a knowledge portal with up-to-date guidance on experimental treatment pathways, eligibility criteria, and regulatory processes tailored to rare disease cases.
FAQ
What is expanded access for experimental therapies?
Expanded access, also called compassionate use, is an FDA program that allows patients with serious or life-threatening conditions to receive investigational drugs outside of clinical trials. The treating physician files an Individual Patient IND using FDA Form 3926.
How long does the expanded access process take?
The standard FDA review period is 30 days for non-emergency requests. The full process from initial evaluation to first treatment dose typically takes 4–5 months.
Can the FDA force a manufacturer to provide an experimental drug?
No. Drug manufacturers decide voluntarily whether to supply investigational drugs. The FDA cannot compel provision, which makes a strong, patient-specific rationale critical when approaching manufacturers.
What is the difference between a clinical trial and expanded access?
Clinical trials collect research data under strict protocols. Expanded access focuses on treating an individual patient. A patient who does not qualify for a trial may still qualify for expanded access using the same drug.
How do I find legitimate experimental therapies for a rare genetic disease?
Start with ClinicalTrials.gov, which lists all registered U.S. studies. Use plain-English eligibility tools to interpret criteria, and consider platforms like Hopeatrarelabs that screen therapies against patient-specific disease models for a more targeted search.