
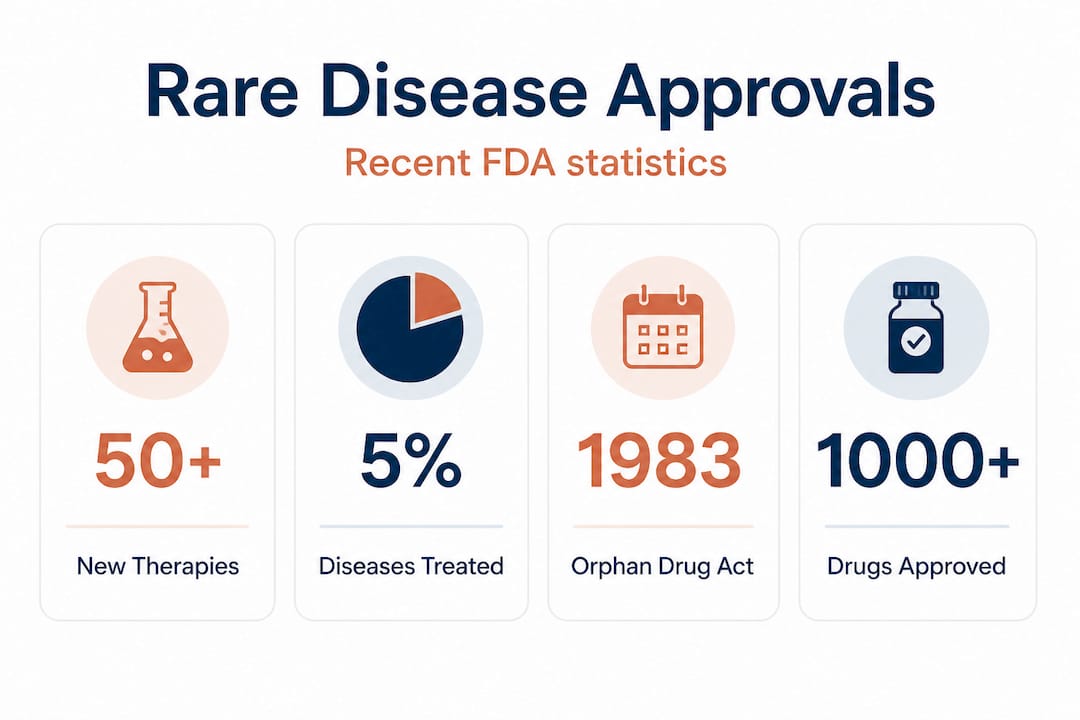
Over 10,000 rare diseases exist, yet only about 5% have an FDA-approved treatment, leaving roughly 30 million Americans without targeted options. For families navigating an ultra-rare diagnosis, that number is not a statistic. It's a reality that shapes every doctor's appointment, every research search, every sleepless night. But the landscape is shifting. A combination of regulatory innovation, flexible trial design, and new individualized therapy frameworks is opening doors that didn't exist a decade ago. This guide explains exactly how FDA approval works for rare and ultra-rare diseases, what's changed recently, and how patients, families, and researchers can use these pathways effectively.
Table of Contents
- What makes a disease 'rare' and why treatments are limited
- FDA pathways: How orphan drugs get approved for rare and ultra-rare diseases
- Breakthrough frameworks for ultra-rare genetic diseases
- Real-world impact: What FDA-approved drugs mean for patients and researchers
- What most guides miss: The double-edged reality of rare disease drug approvals
- Explore your options with RareLabs
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Few rare diseases treated | Only about 5% of rare diseases have FDA-approved drugs despite over 10,000 known conditions. |
| FDA incentives drive innovation | The Orphan Drug Act offers exclusivity, tax credits, and fee waivers to encourage new therapies. |
| Flexible approval for ultra-rares | New frameworks allow single trials, biomarkers, and case data for conditions with tiny patient numbers. |
| Recent progress, but big gaps | FDA approvals are increasing, especially for genetic diseases, but most rare conditions remain unmet. |
| Resources for families exist | Platforms like RareLabs and expanded access help patients discover available and developing treatments. |
What makes a disease 'rare' and why treatments are limited
The FDA defines a rare disease as one affecting fewer than 200,000 people in the United States at any given time. That definition might seem straightforward, but its implications for drug development are enormous. When the potential patient pool is small, the commercial math rarely works out. Companies face the same development costs as mainstream drugs but can only serve a fraction of the market, making profitability uncertain at best.
Before 1983, this economic reality meant that rare diseases were largely ignored by the pharmaceutical industry. The Orphan Drug Act changed that by creating a set of powerful incentives: seven years of market exclusivity after approval, substantial tax credits on clinical trial costs, and a waiver of FDA user fees that can otherwise run into millions of dollars. The law directly jumpstarted rare disease drug development and led to hundreds of approvals that would not have occurred otherwise.
Even with these incentives, the numbers remain stark:
| Category | Estimated figure |
|---|---|
| Total recognized rare diseases | ~10,000 |
| Rare diseases with FDA-approved drugs | ~5% (~500) |
| Americans living with a rare disease | ~30 million |
| Rare diseases that are genetic in origin | ~80% |
The core barriers to developing treatments include:
- Small patient populations that make recruiting for clinical trials slow and expensive
- Lack of natural history data, meaning scientists often don't know how a disease progresses without treatment
- Limited biomarkers to measure whether a drug is actually working
- High per-patient R&D costs spread across fewer individuals than for common diseases
- Fragmented patient communities scattered across the globe, making coordination difficult
Understanding these structural challenges is the first step toward recognizing why overcoming rare disease challenges requires more than just good science. It demands regulatory creativity, patient engagement, and a willingness to redefine what constitutes sufficient evidence.
Statistic callout: Approximately 80% of rare diseases have a genetic root cause, which makes genomic sequencing and gene-level interventions especially relevant for this community.
FDA pathways: How orphan drugs get approved for rare and ultra-rare diseases
Knowing why treatment gaps exist helps, but the more actionable knowledge for families and researchers is understanding how drugs can actually reach approval. The FDA has built a layered system specifically for rare disease drug development, and each layer matters.
Orphan Drug Designation (ODD) is the starting point. When a drug developer applies for and receives ODD, they gain access to the full suite of Orphan Drug Act incentives: tax credits, fee waivers, and the seven-year exclusivity window. Crucially, ODD is granted before clinical trials are complete. It signals that the FDA recognizes the drug is intended for a rare condition and provides development support from the start.
Beyond ODD, the FDA has developed more flexible approaches to evidence as the science has evolved. The Rare Disease Evidence Principles (RDEP), launched in recent years, formally recognize that FDA evidence standards for ultra-rare populations may need to differ from those used in large-scale trials. RDEP allows developers to lean on single pivotal trials, natural history controls, biomarker data, and mechanistic evidence in place of the traditional two-trial gold standard.
Here's a comparison of standard versus rare disease approval approaches:
| Feature | Standard drug approval | Orphan/rare disease approval |
|---|---|---|
| Required pivotal trials | Usually two | Can be one with strong supporting data |
| Patient population in trials | Hundreds to thousands | Dozens to hundreds |
| Surrogate endpoint use | Less common | More common and accepted |
| Natural history controls | Rarely accepted | Accepted with justification |
| FDA engagement during development | Standard | Enhanced, more collaborative |
The FDA's accelerated approval pathway is another critical mechanism. When a drug demonstrates a meaningful effect on a biomarker or surrogate endpoint likely to predict clinical benefit, the FDA can grant conditional approval. The company then continues confirming clinical benefit in a post-market study. This approach has been used effectively for several rare disease therapies, shaving years off the time from discovery to patient access.
- Apply for Orphan Drug Designation early in development
- Engage the FDA for pre-IND (Investigational New Drug) meetings to align on evidence expectations
- Design trials that reflect the patient population's actual size and characteristics
- Consider surrogate endpoints or biomarkers validated through natural history studies
- Use rolling review to submit completed sections of your application while the rest is still in development
Pro Tip: Families and patient advocacy groups can directly petition the FDA and engage with Rare Disease Advocates at the agency. Many successful drug approvals have been accelerated by organized patient community involvement providing natural history data.
For families trying to make sense of this system, finding rare disease treatments is a useful resource to understand what options exist at each stage of a drug's development. Understanding whether a therapy is in a Phase 1 trial, has Breakthrough Therapy designation, or is under expanded access tells you a great deal about realistic timelines.
The FDA also offers rare disease incentives that extend beyond the Orphan Drug Act itself, including Breakthrough Therapy Designation for drugs showing early substantial improvement, Fast Track for serious conditions, and Priority Review for drugs with significant advantages over existing options. Layering these designations is a recognized strategy in rare disease drug development, and personalized rare disease therapies are increasingly benefiting from exactly this combination.
Breakthrough frameworks for ultra-rare genetic diseases
Even the most flexible traditional FDA pathways can struggle when a disease affects only a few dozen or a few hundred people worldwide. Classic randomized controlled trials become logistically impossible. You cannot randomize 15 patients into two groups, add a placebo arm, and generate statistically robust data. The FDA recognized this, and the response has been genuinely innovative.
The Plausible Mechanism Pathway and related frameworks for individualized therapies represent a major conceptual shift. Under these approaches, evidence that a drug corrects the known biological malfunction causing a disease, even without a large clinical trial, can support approval or investigational use. The logic is straightforward: if we know precisely what a genetic mutation does to a cellular process, and we have a therapy that demonstrably corrects that process in patient cells and animal models, waiting for a 200-person trial may cost lives.
Key features of these ultra-rare frameworks include:
- Single-arm trials with natural history or external controls replacing placebo groups
- N-of-1 or very small series case reports accepted as primary evidence when population size makes group studies impossible
- Mechanistic data from laboratory models, including patient-derived induced pluripotent stem cells (iPSCs), used as supporting evidence
- Biomarker endpoints tied directly to the disease mechanism rather than subjective clinical outcomes
- Genome editing and custom RNA therapies evaluated under adaptive trial designs that can evolve as data accumulates
"Ultra-rare frameworks including the Plausible Mechanism Pathway allow individualized genome or RNA-based therapies to advance using single-arm trials and natural history controls where traditional methods are not feasible."
For families whose child has a disease affecting fewer than 100 people in the world, these frameworks are not abstract regulatory theory. They are the realistic path to a therapy. The five-step process for ultra-rare genetic disease intervention broadly follows this structure: identify the causal genetic variant, validate the target in patient-derived cells, build a disease model that replicates the biology, test an intervention in that model, and confirm measurable biological benefit. Each step produces evidence that feeds into a regulatory case.
Research teams exploring genetic therapies for ultra-rare diseases are increasingly using CRISPR gene editing and antisense oligonucleotides (ASOs) within this framework. ASOs in particular can be custom-designed for a specific mutation in a matter of months, and the FDA has approved N-of-1 ASO therapies for individual patients, a landmark moment in personalized medicine. If you want to understand what those approaches look like technically, resources on gene therapy for ultra-rare disorders lay out the mechanisms and current evidence in accessible terms.

Pro Tip: If your disease affects fewer than 1,000 patients worldwide, ask your specialist whether a natural history registry exists. Contributing data to such a registry actively builds the evidence base that the FDA will need to evaluate any future therapy for your condition.
Real-world impact: What FDA-approved drugs mean for patients and researchers
The frameworks above are only valuable if they translate into actual approved drugs reaching actual patients. Recent data shows that translation is increasingly happening.
FDA rare disease approvals from 2021 through 2026 include more than 50 novel therapies, spanning conditions from TK2 deficiency to WHIM syndrome. In 2025, orphan drugs represented roughly 50% of all novel drug approvals from the FDA, a milestone that reflects how deeply rare disease development has reshaped the pharmaceutical pipeline.
| Recent notable approvals | Condition | Mechanism |
|---|---|---|
| Kygevvi | TK2 mitochondrial myopathy | Nucleoside substrate enhancement |
| Xolremdi | WHIM syndrome | CXCR4 antagonist |
| Casgevy | Sickle cell disease and beta thalassemia | CRISPR gene editing |
| Skysona | Cerebral adrenoleukodystrophy | Lentiviral gene therapy |
For patients and families, these approvals matter in several concrete ways:
- Direct access: An approved drug can be prescribed, covered by insurance, and administered in clinical settings without navigating complex access programs.
- Validated mechanism: Approval means the FDA found credible evidence that the drug works through its intended pathway, reducing uncertainty.
- Expanded access before approval: The FDA's compassionate use program allows seriously ill patients to access investigational drugs before final approval when no alternatives exist.
- Research momentum: Each approval generates data, attention, and funding that often benefits adjacent conditions.
For researchers and biopharma companies, the rare disease approval statistics tell a story of rising ROI alongside rising scientific complexity. The incentive structure works. But the hurdles around pricing, post-market commitments, and patient access remain genuinely difficult to navigate.
Work on identifying the right therapeutic targets for rare diseases is where scientific and commercial interest converges. Picking the right target, validating it early with patient-derived models, and building an evidence package that aligns with current FDA frameworks is the formula that moves drugs forward fastest. For teams considering gene therapy specifically, a thorough gene therapy evaluation guide can clarify what questions to ask before committing to a development pathway.
What most guides miss: The double-edged reality of rare disease drug approvals
Here's something most celebratory summaries of rare disease progress leave out: approvals are remarkable and genuinely transformative, but they don't resolve the deeper inequity in rare disease medicine.
Despite more than 1,000 orphan drugs approved since 1983, 95% of rare diseases still have no approved treatment. That gap is not closing as fast as the growing orphan drug pipeline might suggest, because most approved drugs cluster around conditions with larger patient populations within the rare disease category. Conditions affecting a few hundred or a few thousand people globally often remain behind.
There's also a tradeoff embedded in the Orphan Drug Act's exclusivity provision that rarely gets discussed openly. Seven years of market exclusivity enables profitability, but it also keeps prices high during those years. Some approved orphan drugs cost hundreds of thousands of dollars annually. For families without robust insurance or in countries where those drugs aren't available, an FDA approval can feel more like a headline than a solution.
The uncomfortable truth is that regulatory approval and actual patient access are not the same thing. Families should not treat "FDA-approved drug exists" as the finish line. They should understand expanded access, compassionate use, clinical trial enrollment, and international access programs as part of their toolkit, even while pushing for formal approval. Proactively addressing barriers in rare disease research means families and advocacy groups need to engage the system at every level, not just wait for it to produce answers.
The most meaningful progress tends to come when patient communities, researchers, and regulators collaborate from day one. Not when each waits for the other to act. The RDEP and individualized therapy frameworks exist partly because patient advocates pushed for them. That influence is real, and it should be used.
Explore your options with RareLabs
If you're a patient, family member, or research partner looking for concrete next steps after learning about these pathways, you don't have to navigate this landscape alone.
RareLabs builds patient-specific disease models from your own cells, using iPSC technology and CRISPR gene editing to test thousands of FDA-approved drugs, custom ASOs, and gene therapy options in parallel. Our platform is designed specifically for ultra-rare and undiagnosed conditions where no approved treatment exists yet. You can explore treatment possibilities, access the latest regulatory developments, and get guidance on expanded access and clinical trial options through our rare disease treatment knowledge center. When you're ready to take a personalized next step, visit hopeatrarelabs.com to learn how we work with families and research teams to find answers faster.
Frequently asked questions
What is the Orphan Drug Act and how does it help rare diseases?
The Orphan Drug Act provides drug developers with seven years of market exclusivity, significant tax credits on clinical trial costs, and waived FDA user fees to encourage investment in treatments for diseases affecting fewer than 200,000 people in the US.
How can families find out if there's an FDA-approved drug for their ultra-rare disease?
Families can search Drugs@FDA directly or use resources like the RareLabs knowledge center; if no approved option exists, expanded access programs and compassionate use may allow access to investigational therapies before formal approval.
Why do so few rare diseases have FDA-approved drugs?
High research costs combined with very small patient populations make investment difficult; the result is that only ~5% of rare diseases have approved treatments despite decades of regulatory incentives designed to address exactly this problem.
What are the new FDA frameworks for ultra-rare diseases?
Programs like RDEP and the Plausible Mechanism Pathway allow developers to use mechanistic data, biomarkers, and single-arm trials as valid evidence when traditional clinical trials aren't possible due to extremely small patient populations.
How do researchers and companies take advantage of orphan drug programs?
The most effective strategy is to apply early for ODD and schedule FDA pre-IND meetings before committing to a full trial design; early engagement aligns development plans with the FDA's current evidence standards and maximizes access to available incentives.