
Traditional drug development takes 10 to 15 years. For a child with an ultra-rare genetic disease, that timeline is not a statistic — it is a life. The good news is that the landscape is shifting faster than most families realize. New regulatory frameworks, AI-driven screening tools, and patient-led data networks are creating genuine ways to accelerate rare disease therapy development, compressing timelines that once felt immovable. This guide breaks down every major lever available to families, researchers, and biopharma partners right now — not the standard overview, but the specific programs, tools, and decisions that actually move things forward.
Table of Contents
- Ways to accelerate rare disease therapy through regulatory advances
- Leveraging technology innovations to shorten therapy development timelines
- Harnessing data networks and patient partnerships to reduce trial timelines
- Comparing pathways to accelerated rare disease therapies
- Practical steps to accelerate therapy development for patients and researchers
- Why speed alone is not the whole answer
- RareLabs: Patient-specific models built for therapy acceleration
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Regulatory flexibility | FDA frameworks allow approval of therapies based on small patient data sets for ultra-rare diseases. |
| Technology impact | AI and genome editing programs greatly reduce diagnosis and development timelines. |
| Collaboration accelerates | Patient registries and data sharing networks cut trial recruitment time significantly. |
| Family engagement matters | Families can drive research progress by contributing data and partnering with biopharma. |
| Practical steps | Early FDA engagement and participation in acceleration programs shorten therapy pathways. |
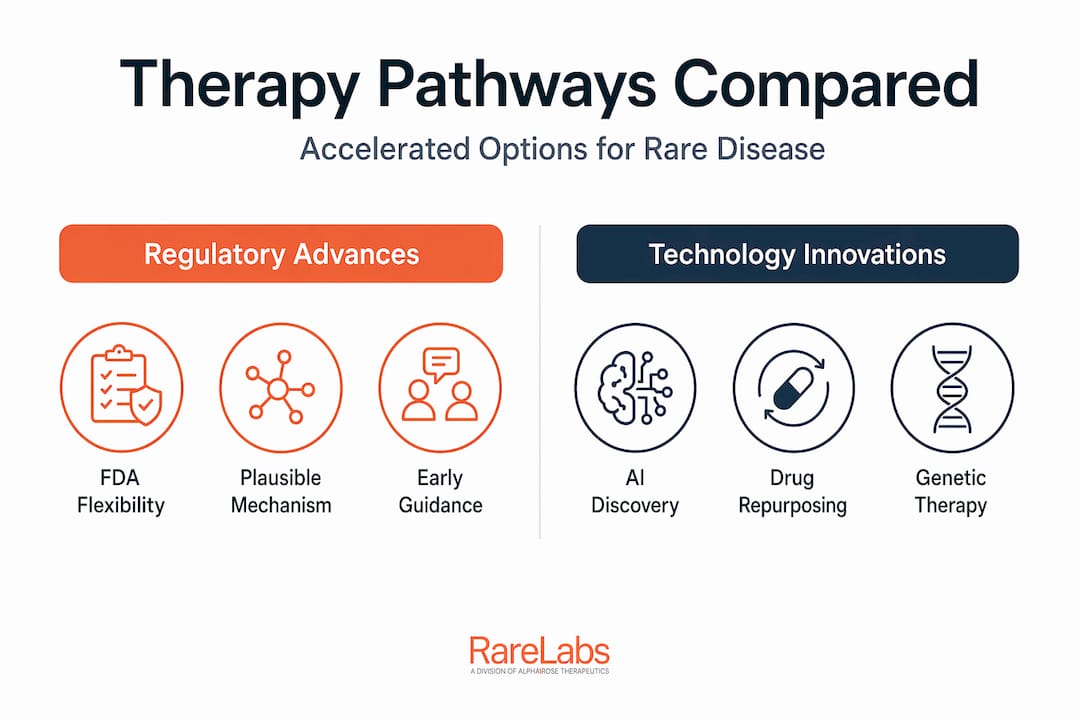
Ways to accelerate rare disease therapy through regulatory advances
The FDA has made a genuine pivot in how it thinks about ultra-rare diseases. For conditions affecting fewer than 1,000 people worldwide, traditional randomized controlled trials are often impossible. You simply cannot recruit 200 patients when only 40 exist. Regulatory frameworks now reflect that reality.
The most significant development is the FDA Plausible Mechanism Framework, which allows approval of individualized therapies using evidence from 1 to 5 patients. The framework does not lower the scientific bar — it changes the evidentiary structure. Instead of requiring statistical power from large cohorts, it requires a strong mechanistic rationale: proof that a therapy targets the root genetic cause in a scientifically defensible way. This opens the door to personalized rare disease treatments that would never survive the traditional approval process.
Alongside this, the FDA's ARC Program coordinates efforts across disease areas and develops surrogate endpoints that can shorten trial durations by 2 to 5 years. Surrogate endpoints are measurable biomarkers — enzyme activity levels, protein expression, or neurological function scores — that stand in for harder-to-measure clinical outcomes. Instead of waiting years to see whether a child walks independently, a trial can use a validated biomarker that predicts that outcome.
Here is what this means practically for families and research teams:
- Single pivotal trial model: The FDA now allows one well-designed pivotal trial plus real-world evidence (RWE) to support approval, instead of requiring two independent trials. This matters enormously for conditions where patient numbers simply cannot support duplicate trials.
- Surrogate endpoint negotiation: Sponsors can submit early to the FDA and negotiate which endpoints qualify, avoiding costly redesigns mid-trial.
- Accelerated Approval pathway: Available when a surrogate endpoint is reasonably likely to predict clinical benefit, often cutting years from the timeline.
- Orphan Drug Designation: Provides seven years of market exclusivity, tax credits, and waived application fees, reducing financial barriers that kill rare disease programs early.
"The FDA's shift toward mechanistic evidence and surrogate endpoints is not a regulatory shortcut. It is an acknowledgment that the scientific question in ultra-rare disease is fundamentally different: not 'does this work in most people?' but 'does this correct the biological defect in this patient?'"
Understanding FDA rare disease drug guidance early in the process can prevent years of avoidable delay. Regulatory strategy is not just a legal exercise — it is a core scientific decision.
Leveraging technology innovations to shorten therapy development timelines
Regulatory flexibility creates the opening. Technology fills it. The combination of AI-driven drug discovery, digital patient modeling, and genome editing programs is compressing timelines in ways that were not possible five years ago.

Drug repurposing cuts development time from 15 years to 7.5 years and reduces costs by 80% using AI screening. That is not a marginal improvement — it is a structural change in how therapy discovery works for rare conditions. Instead of developing a new molecule from scratch, AI platforms screen thousands of FDA-approved drugs against a patient's specific mutation, identifying candidates with known safety profiles that might correct the underlying defect. The safety data already exists. The manufacturing process already exists. You are simply redirecting a known tool toward a new target.
ARPA-H programs RAPID, MATRIX, and THRIVE each address a different bottleneck: faster diagnosis, AI-powered drug repurposing, and genome editing program expansion respectively. THRIVE, in particular, is building infrastructure for bespoke genome editing therapies targeting mutations that are too rare and too unique for traditional drug development economics.
Here is a practical sequence for how these technologies compress the path from diagnosis to therapy:
- Genetic sequencing and AI-powered interpretation identify the causal mutation, often reducing diagnostic odysseys from years to weeks.
- Digital patient profiles and in silico modeling predict which existing drugs or compounds might interact with the mutation, generating a ranked list of candidates before any lab work begins.
- Patient-derived cell models (including iPSC-derived neurons, cardiomyocytes, or liver cells) validate those predictions in a biologically accurate system, filtering candidates fast.
- Parallel drug screening tests multiple candidates simultaneously rather than sequentially, cutting months out of the process.
- Regulatory submission with mechanistic and in vitro evidence supporting a trial design built around the data already generated.
Exploring genetic therapy options alongside drug repurposing maximizes the probability of finding something that works, because the two approaches are often complementary rather than competing. Meanwhile, advances in gene editing for rare diseases mean that even single-patient mutations with no existing drug match have a viable path forward.
Pro Tip: Engage with AI-driven drug screening platforms as early as possible in the process, even before a formal research program is established. The data generated during screening often informs trial design, FDA submissions, and biopharma partnership conversations simultaneously.
Harnessing data networks and patient partnerships to reduce trial timelines
Even the best regulatory framework and the most capable AI platform hit the same wall: not enough patients, not enough data. This is where patient communities and coordinated data networks become a genuine scientific asset — not just an emotional one.
NORD Rare Disease Centers of Excellence reduce trial recruitment time by 50 to 70% through shared registries and disease-specific expertise. These centers link patients across geographies, standardize clinical assessments, and maintain longitudinal data that would otherwise never exist. For researchers, this means the patient population needed for a trial is not scattered and invisible — it is organized, characterized, and trial-ready.
RDCRN and RDCA-DAP platforms enable federated data sharing across global registries, avoiding data silos that delay therapies by 3 to 5 years. Federated queries allow researchers to ask questions across multiple registries without moving patient data, which protects privacy while enabling the kind of aggregate analysis that drives trial design decisions.
Here is what families can do directly to contribute to and benefit from these networks:
- Register with disease-specific patient registries as early as possible, even before a formal diagnosis if the suspected condition has an existing registry.
- Document natural history data including functional assessments, biomarker measurements, and clinical milestones — this data becomes the foundation for trial eligibility criteria.
- Connect with NORD Centers for multidisciplinary care that simultaneously generates trial-quality clinical data.
- Participate in patient advocacy networks that communicate directly with biopharma companies, flagging unmet needs and demonstrating organized patient communities that reduce the business risk of rare disease programs.
Pro Tip: Natural history data collected by families, when standardized and registry-linked, is often the single most persuasive asset for attracting a biopharma partner to a rare disease program. Organized data changes the economics of the decision.
| Data source | What it contributes | Impact on timelines |
|---|---|---|
| NORD Centers | Clinical assessments, biomarkers, linked registries | Reduces recruitment by 50 to 70% |
| RDCRN/RDCA-DAP | Federated global registry queries | Eliminates 3 to 5 year data gaps |
| Patient-led natural history | Functional milestones, treatment responses | Informs trial design before trials begin |
| Disease-specific foundations | Funding, biopharma connections, advocacy | Attracts research programs faster |
Addressing rare disease research challenges systematically — rather than one family at a time — is what makes these networks genuinely powerful for speeding up the genetic disease research process.
Comparing pathways to accelerated rare disease therapies
Not every acceleration pathway fits every disease. Understanding the tradeoffs helps families and researchers prioritize where to invest time and resources.
| Pathway | Typical timeline | Evidence required | Best suited for |
|---|---|---|---|
| Plausible Mechanism Framework | 2 to 4 years | 1 to 5 patients, mechanistic rationale | Single-gene ultra-rare diseases |
| Drug repurposing with AI | 5 to 8 years | In vitro + small cohort | Conditions with druggable mutations |
| Single pivotal trial + RWE | 4 to 7 years | One trial, real-world data supplement | Small but enrollable populations |
| Surrogate endpoint trials | 3 to 6 years | Validated biomarker + clinical correlation | Diseases with measurable biomarkers |
| Tailored genome editing | 3 to 7 years | Preclinical + mechanistic + small cohort | Single-mutation conditions, no drug match |
The FDA permits a single pivotal trial plus real-world evidence, cutting approval time by 1 to 2 years compared to the traditional two-trial model. For researchers designing a program today, this is the default assumption — not an exception to pursue.
Key considerations when selecting a pathway:
- Patient population size determines whether surrogate endpoints or mechanistic frameworks are more feasible.
- Mutation type shapes whether drug repurposing or gene editing is the more direct route.
- Existing natural history data affects how quickly a trial can be designed and launched.
- Regulatory history for similar conditions can inform which evidence package the FDA is most likely to accept.
Reviewing a gene therapy evaluation guide helps map which gene therapy approaches align with the specific mutation and population characteristics you are working with.
Practical steps to accelerate therapy development for patients and researchers
Knowing the frameworks matters less than knowing what to do next. Here is the action sequence that moves a rare disease program from concept to clinical reality.
- Attend a Rare Bootcamp. Patient families attend free Rare Bootcamps to learn research skills and attract biopharma partners. These programs teach families how to build natural history data, understand drug development economics, and present their disease in a way that resonates with research sponsors.
- Join Global Genes RAP. Joining Global Genes Research Acceleration Program helps identify research gaps and connects stakeholders to shorten trial paths. It links families to academic researchers and biopharma partners who might otherwise never encounter the disease.
- Submit a pre-submission request to the FDA. Early FDA engagement lets sponsors shape the trial design, agree on endpoints, and avoid regulatory surprises that routinely add 12 to 18 months to programs.
- Connect with NORD Centers for trial matching. NORD Centers actively match patients with open trials and can provide the clinical infrastructure needed to collect trial-quality data during routine care.
- Build a disease model in parallel. Patient-derived cell models can be developed while regulatory and advocacy work is underway, so the science is ready when partners and funding arrive.
Pro Tip: Start data collection and advocacy simultaneously, not sequentially. Families who wait until they have "enough" data before reaching out to researchers or biopharma partners consistently lose 12 to 24 months they cannot recover.
The research process guide outlines each phase in detail. For families ready to take the next step, exploring genetic therapies alongside drug repurposing creates the widest possible net.
Why speed alone is not the whole answer
Here is the perspective most acceleration guides skip: moving fast without a biologically accurate model of the disease often accelerates failure. We have seen programs rush into trials using cell lines that do not reflect the patient's actual mutation, or repurpose drugs based on pathway logic that did not hold up in patient-derived tissue. The result is faster negative data — which helps the field but does not help the child.
The acceleration methods described in this article work precisely because they start with biological accuracy. The Plausible Mechanism Framework requires mechanistic rigor. AI repurposing is only as good as the disease model it screens against. Surrogate endpoints only shorten trials if they actually reflect what is happening in the patient's cells. Speed and scientific precision are not in tension here — they are mutually dependent.
The families and researchers who move fastest are not the ones chasing the nearest open door. They are the ones who invest early in understanding their specific mutation, building a model that reflects it, and then running every acceleration tool against that model. That sequence is slower at the start and dramatically faster at every step after.
RareLabs: Patient-specific models built for therapy acceleration
If your family is navigating an ultra-rare or undiagnosed genetic disease, the steps above are clearer with a scientific partner who has built infrastructure specifically for this work.
At RareLabs, we create disease models derived from your child's own cells, using iPSCs and CRISPR gene editing to replicate the exact biological defect driving the disease. Against that model, we run parallel screens covering thousands of FDA-approved drugs, custom antisense oligonucleotides (ASOs), and gene therapy candidates simultaneously — not sequentially. Every result is shared transparently, with your physician and your team, in plain language. If you are ready to move from searching to screening, start with RareLabs and we will show you exactly what a personalized therapy search looks like for your specific mutation.
Frequently asked questions
What is the FDA Plausible Mechanism Framework and how does it help ultra-rare disease therapies?
The FDA Plausible Mechanism Framework allows approval of individualized therapies with evidence from 1 to 5 patients, using strong scientific rationale rather than large-cohort statistics. It is specifically designed for conditions where traditional randomized controlled trials are impossible due to patient population size.
How does drug repurposing speed up treatment development for rare diseases?
AI-driven repurposing screens thousands of existing FDA-approved drugs against a patient's mutation, and development time drops from 15 years to 7.5 years while costs fall by 80%. Because safety profiles already exist for these drugs, the path to clinical use is significantly shorter than developing a new compound.
How can patient families contribute to accelerating therapy development?
Families who attend Rare Bootcamps learn how to build natural history data and present their disease to biopharma partners in a format that actually drives research decisions. Organized, standardized patient data is one of the most persuasive assets for attracting a research sponsor to an ultra-rare disease program.
What role do patient registries and real-world data play in speeding rare disease trials?
NORD Rare Disease Centers reduce trial recruitment time by 50 to 70% through coordinated registries and shared clinical expertise. Real-world data collected through these networks also supplements trial evidence, enabling single-trial approval pathways that cut years from the regulatory timeline.