
Transparency in biotech research is defined as the open disclosure of trial data, research methodologies, regulatory decisions, and funding sources to all relevant stakeholders. The role of transparency in biotech research extends beyond ethics. It directly determines whether findings can be reproduced, whether clinicians can trust published results, and whether policymakers can act on accurate evidence. The FDA's 2025 initiative to publish Complete Response Letters in real time and the NIH 2025 Public Access Policy eliminating 12-month embargo periods both signal a structural shift. Biotech research that withholds data does not just risk credibility. It risks patient safety.
How does transparency in biotech research affect integrity and outcomes?
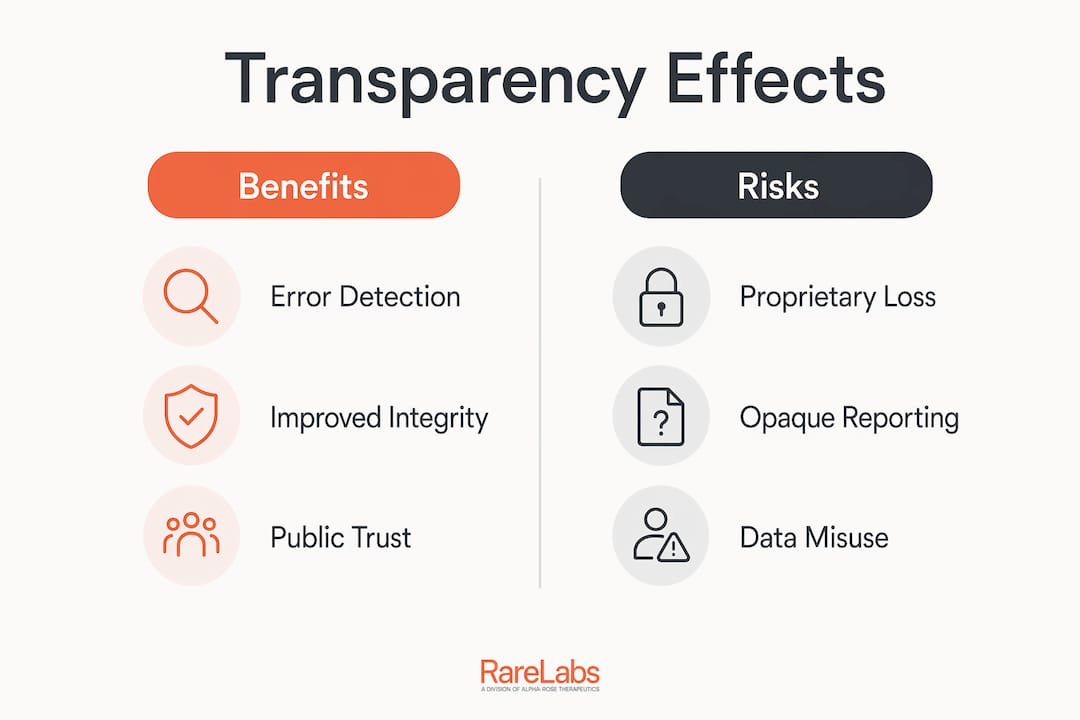
Transparency in biotech research is the single most reliable mechanism for catching errors before they cause harm. When trial results, protocols, and analytical methods are publicly available, independent researchers can verify findings. When they are not, selective reporting fills the gap, and clinicians make decisions based on incomplete pictures.
The FDA has contacted over 2,200 clinical trial sponsors to enforce result disclosure obligations as of april 2026. That scale of outreach reflects how widespread non-disclosure has been, not how rare it is. Outcome reporting bias, where sponsors publish only favorable results, distorts the evidence base that clinicians and investors rely on.

The consequences of opaque reporting are concrete. Drugs that fail in unreported trials may be tested again in new populations, exposing patients to known risks without the benefit of prior negative data. Reproducibility collapses when methods are hidden. Regulatory confidence erodes when agencies cannot verify what sponsors claim.
Transparency in biomedical research also affects investment decisions. Investors who cannot access trial outcomes make capital allocation decisions on incomplete data. That misallocation slows the development of genuinely promising therapies, particularly in rare disease research where patient advocacy depends on accurate public information.
Key practices that support clinical trial transparency include:
- Prospective trial registration on public registries before enrollment begins
- Full results disclosure within 12 months of trial completion, regardless of outcome
- Protocol publication before data collection to prevent post-hoc endpoint changes
- Adverse event reporting that includes negative and null findings, not just statistically significant results
Pro Tip: If you are a researcher submitting trial results, publish your statistical analysis plan before unblinding. This single step eliminates the most common form of selective reporting bias and strengthens your findings' credibility with regulators and peer reviewers.
What does the FDA's real-time CRL publication mean for biotech companies?
The FDA launched real-time publication of Complete Response Letters in july 2025. Over 200 CRLs have been published publicly to date. The goal is direct: prevent biotech companies from issuing investor communications that mischaracterize regulatory rejections.
Before this policy, a company could receive a CRL citing major safety deficiencies and issue a press release describing the outcome as a "minor labeling issue." Investors had no independent way to verify that framing. Real-time CRL publication removes that information asymmetry entirely.
The implications for biotech companies are significant and require immediate operational adjustments:
- Align all investor communications with CRL language. Any gap between what the FDA documents and what a company states publicly creates securities litigation exposure.
- Establish internal review protocols so that legal, regulatory, and communications teams review CRL content before any public statement is issued.
- Prepare for redaction disputes. Companies may request redactions of proprietary manufacturing data, but the FDA's default is disclosure.
- Audit past communications. Companies with pending applications should review prior investor statements for consistency with any CRL content that may become public.
- Train investor relations teams on regulatory terminology so that CRL findings are described accurately, not minimized.
"Radical transparency through FDA CRL publication is raising accountability and professionalism in biotech communications. Companies that treat this as a compliance burden miss the point. Those that treat it as a trust-building opportunity gain a durable advantage with regulators and investors alike."
Accountability in biotech does not stop at the CRL. Firms view regulatory meeting details as extremely proprietary, and many resist releasing FDA meeting minutes even when transparency policies are in place. That resistance creates a partial transparency problem: public CRLs without the meeting context that explains them can still mislead. The policy is a strong step forward, but the gap between published letters and unpublished meeting discussions remains a live issue for researchers and policymakers tracking regulatory accountability.
How can biotech balance transparency with proprietary information?
Open disclosure and intellectual property protection are not mutually exclusive. The EU pharma package demonstrates this clearly. Its transparency framework focuses on structured metadata disclosures covering patents, licensing status, and transfer restrictions, rather than requiring companies to reveal trade secrets or proprietary manufacturing processes.
This distinction matters for follow-on research. When a platform technology's patent family and licensing terms are publicly known, academic researchers and smaller biotech firms can assess whether building on that technology is legally viable. When that information is hidden, researchers waste resources pursuing paths that are already locked up, or they avoid entire research areas out of uncertainty.
| Transparency approach | What it discloses | What it protects |
|---|---|---|
| Metadata disclosure | Patent families, licensing status, transfer restrictions | Manufacturing processes, formulation details |
| Protocol publication | Study design, endpoints, analysis plan | Proprietary assay methods |
| Results registration | Outcomes, adverse events, statistical results | Unpublished compound libraries |
| Plain-language summaries | Findings in accessible language | Raw datasets with identifiable patient data |
Opaque licensing creates a specific harm in rare disease research. When a gene therapy platform is encumbered by undisclosed licensing restrictions, patient-specific modeling programs cannot access the tools they need. Hopeatrarelabs works within this reality daily, building personalized disease models using iPSCs and CRISPR gene editing while navigating the licensing constraints that affect which therapeutic approaches are accessible.
Pro Tip: When publishing research that uses a licensed platform technology, disclose the licensing category (exclusive, non-exclusive, field-of-use restricted) in your methods section. This single disclosure saves downstream researchers significant time and prevents wasted replication attempts.
What are FAIR data principles and why do they matter in biotech?
Open data in research means more than making files available. Data that is technically public but practically inaccessible does not advance science. The FAIR principles, standing for Findability, Accessibility, Interoperability, and Reuse, define the standard that makes transparency functional rather than symbolic.
Open Pharma advocates for FAIR data principles and plain-language summaries as the operational definition of genuine transparency. A dataset locked behind a proprietary format, or a clinical summary written in regulatory jargon, fails the transparency test even if it is technically disclosed. Accessibility requires that data be retrievable by researchers using standard tools. Interoperability requires that datasets can be combined across studies. Reuse requires that licensing terms permit secondary analysis.
The benefits of research transparency built on FAIR principles include:
- Reproducibility: Independent teams can rerun analyses using the same datasets and code, confirming or challenging original findings
- Meta-analysis capability: Interoperable datasets from multiple trials can be pooled, increasing statistical power for rare disease research where individual trial sizes are small
- Patient involvement tracking: Plain-language summaries allow patients and families to assess whether research reflects their priorities and experiences
- Policy relevance: Policymakers without deep technical training can engage with accessible summaries rather than relying solely on expert interpretation
Transparency policies often remain aspirational without enforceable monitoring mechanisms. The reproducibility crisis in biotech research continues despite existing mandates. Public-facing compliance monitoring, not just policy statements, is what closes the gap between what institutions promise and what researchers actually deliver. For clinicians tracking medical research updates, the difference between aspirational and enforced transparency is the difference between reliable and unreliable evidence.
The NIH 2025 Public Access Policy removes the 12-month embargo that previously delayed public access to federally funded research. That change accelerates the availability of findings for researchers working on time-sensitive rare disease cases. The policy is a meaningful structural improvement, though implementation gaps remain significant, particularly for smaller research groups that lack the infrastructure to meet open-access requirements affordably.
Key Takeaways
Transparency in biotech research requires enforceable disclosure of trial results, regulatory communications, and FAIR-compliant data to produce reproducible science and trustworthy outcomes.
| Point | Details |
|---|---|
| Clinical trial disclosure | The FDA contacted over 2,200 sponsors to enforce result publication and reduce outcome reporting bias. |
| Real-time CRL publication | Over 200 Complete Response Letters are now public, requiring companies to align investor communications with regulatory findings. |
| Metadata over trade secrets | EU frameworks show that patent and licensing disclosures enable follow-on research without exposing proprietary processes. |
| FAIR principles are the standard | Findability, Accessibility, Interoperability, and Reuse define functional transparency beyond raw data availability. |
| Enforcement closes the gap | Aspirational policies without compliance monitoring fail to improve reproducibility or data sharing in practice. |
Why I think the transparency debate is asking the wrong question
Most discussions about transparency in biotech frame it as a tension between openness and protection. Researchers worry about being scooped. Companies worry about litigation. Regulators worry about proprietary manufacturing details. All of those concerns are real. But the framing misses what I have seen matter most in practice.
The question is not how much to disclose. The question is whether the disclosure is usable. A 400-page clinical study report published as a scanned PDF satisfies a legal transparency requirement and helps no one. A structured dataset with a clear data dictionary, published with the analysis code, advances science immediately. The NIH Public Access Policy and the FDA's CRL initiative are both steps in the right direction. But neither addresses the usability gap that makes so much "transparent" research practically opaque.
What I recommend for researchers and institutions is simple. Treat transparency as a design principle, not a compliance checkbox. Build your data management plan before you collect data, not after. Write your plain-language summary before you write your abstract. Register your trial before you enroll your first patient. These are not burdensome additions. They are the practices that make your research worth citing, replicating, and building on.
For policymakers, the lesson from the enforcement gap is equally clear. Mandates without monitoring are wishes. The FDA's outreach to over 2,200 sponsors shows what active enforcement looks like. That model, applied to data sharing and FAIR compliance, would do more for research integrity than any number of aspirational policy statements.
— John
Hopeatrarelabs and transparent rare disease research
Hopeatrarelabs builds its entire research model on the principle that transparency is not optional in rare disease work. When a patient has an ultra-rare or undiagnosed genetic disease, every data point matters, and every finding must be accessible to the physicians and families who need it.
The RareLabs Knowledge platform gives researchers, clinicians, and policymakers searchable access to rare disease research and treatment data in one place. It reflects the same commitment to open data in research that the FDA and NIH are now enforcing at the regulatory level. If you work in rare disease research or advise patients navigating undiagnosed conditions, the platform offers a direct path to the transparent, up-to-date information that drives better decisions.
FAQ
What is transparency in biotech research?
Transparency in biotech research is the open disclosure of trial protocols, results, regulatory decisions, and data in formats that stakeholders can access and use. It covers clinical trial registration, results publication, and FAIR-compliant data sharing.
Why did the FDA contact over 2,200 clinical trial sponsors?
The FDA reached out to enforce result disclosure obligations and reduce outcome reporting bias. Sponsors are required to publish trial outcomes publicly, and many had not complied with existing mandates.
What are Complete Response Letters and why are they now public?
Complete Response Letters are FDA communications explaining why a drug application was not approved. The FDA began publishing them in real time in july 2025 to prevent companies from mischaracterizing regulatory rejections in investor communications.
How do FAIR principles improve research transparency?
FAIR principles require that research data be Findable, Accessible, Interoperable, and Reusable. They move transparency from technical availability to practical usability, enabling reproducibility and secondary analysis across studies.
How does the NIH 2025 Public Access Policy change research access?
The NIH 2025 Public Access Policy eliminates the previous 12-month embargo on publicly funded research, requiring immediate open access to findings. This accelerates the availability of new evidence for researchers and clinicians working on time-sensitive cases.