
Undiagnosed disease models are patient-specific, functional platforms built from a patient's own cells that enable precise diagnosis and personalized therapy development for rare and mysterious genetic conditions. The Undiagnosed Diseases Network at NIH has evaluated over 3,500 patients, delivering diagnoses for more than 1,000 rare or unknown conditions through multidisciplinary approaches. That scale of impact is only possible because these models do what traditional clinical pathways cannot: they translate ambiguous genomic data into functional, testable biology. Technologies like induced pluripotent stem cells (iPSCs), patient-derived organoids, and CRISPR genome editing now form the backbone of this work. Understanding why undiagnosed disease models are important begins with recognizing that roughly 6% of rare disease patients remain without a diagnosis, generating fragmented, costly, and emotionally exhausting care pathways that these models are uniquely positioned to resolve.
Why undiagnosed disease models are important for faster diagnosis
The diagnostic odyssey is one of medicine's most persistent failures. Patients with undiagnosed diseases wait over 4 years, sometimes up to 7 years, before receiving a diagnosis. Structured genomic programs that incorporate disease modeling compress that timeline to 12 to 18 months. That is not a marginal improvement. It represents years of unnecessary suffering, misapplied treatments, and repeated hospitalizations eliminated for patients and families who have often exhausted every conventional option.
The mechanism behind this acceleration is systematic. Genomic sequencing generates enormous variant data, but raw sequence data alone rarely delivers a diagnosis. Disease models provide the functional layer: a patient's iPSC-derived neurons or organoids can be observed directly to confirm whether a candidate variant actually disrupts cellular behavior. This closes the gap between genetic suspicion and clinical certainty. Systematically reanalyzing previously unsolved genomic data increases diagnostic yield by over 17%, which means cases once labeled "unsolvable" become diagnoses as scientific knowledge evolves.
The downstream effects of faster diagnosis extend well beyond the individual patient. A confirmed diagnosis enables genetic counseling for families, guides therapy selection, and ends the cycle of trial-and-error treatments that drain both patients and health systems. For pediatric patients in particular, early genomic testing prevents years of inconclusive specialist visits that delay developmental interventions. The undiagnosed disease significance here is not abstract. It is measured in years of life quality restored.
Pro Tip: If a patient's genomic case has been marked "unsolved," request formal reanalysis every 12 to 18 months. Evolving variant databases and new disease gene discoveries regularly convert previously negative cases into confirmed diagnoses.
| Diagnostic approach | Typical timeline | Key advantage |
|---|---|---|
| Traditional clinical pathway | 4 to 7 years | Broad symptom coverage |
| Structured genomic program | 12 to 18 months | Systematic variant interpretation |
| Genomic reanalysis of prior cases | Variable, often within months | Converts unsolved cases using updated science |
| iPSC or organoid functional modeling | Weeks to months post-sequencing | Confirms variant pathogenicity directly |
How patient-derived models compare to traditional disease models
Traditional animal models and 2D cell cultures have driven decades of biomedical discovery, but they carry a fundamental limitation in rare disease research: they do not replicate human-specific pathogenic mechanisms with sufficient fidelity. A mouse model of a rare genetic condition may show some phenotypic overlap, yet the underlying cellular biology often diverges enough to produce misleading drug response data. For ultra-rare diseases where only a handful of patients exist globally, that translational gap is not a minor inconvenience. It is a research dead end.

Patient-derived organoids and iPSC models solve this directly. Patient-derived organoids bypass limitations of traditional animal models by enabling functional interrogation of rare variants in a human-relevant 3D context. This matters because 3D architecture replicates tissue-level interactions that flat 2D cultures miss entirely. A patient's iPSC-derived cardiac organoid, for example, will exhibit the same ion channel dysfunction causing their arrhythmia, something a rodent model may not reproduce at all.
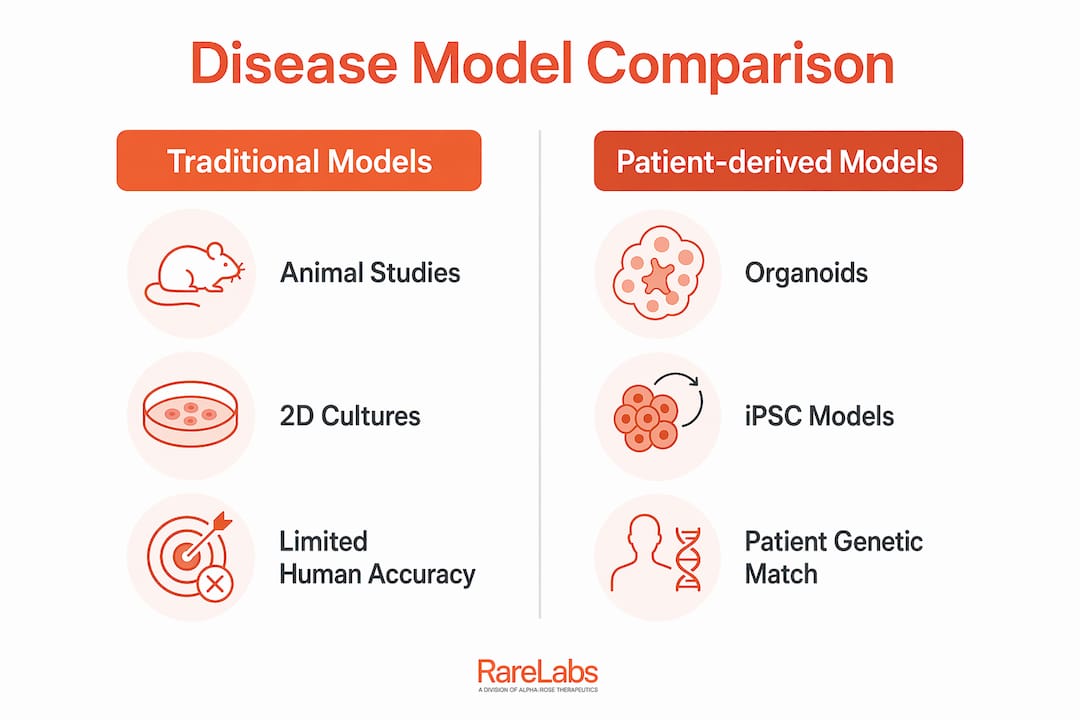
CRISPR genome editing amplifies the power of these platforms further. The combination of CRISPR with iPSC-derived organoids enables precise gene-function studies linking genotype to phenotype and supports personalized therapeutic strategies. Researchers can introduce or correct a specific variant in an organoid, observe the functional consequence, and then screen candidate therapies against the corrected versus uncorrected tissue. This creates a closed-loop system for disease modeling that no animal model can replicate at this resolution.
Key distinctions between model types:
- Traditional animal models: Accessible and well-characterized, but limited by species-specific biology and poor translation to human rare disease phenotypes.
- 2D cell cultures: Fast and cost-effective for initial screening, but lack the multicellular architecture needed to model tissue-level disease.
- Patient-derived iPSCs: Genetically identical to the patient, reprogrammable into virtually any cell type, and suitable for long-term study.
- Patient-derived organoids: Provide 3D multicellular structure, self-organization, and tissue-specific function closest to in vivo human biology.
- CRISPR-edited organoid systems: Enable isogenic comparisons, high-throughput drug screens, and direct genotype-to-phenotype validation.
For researchers studying ultra-rare conditions, the benefits of disease models built from patient cells are not theoretical. They are the difference between a publishable hypothesis and a clinically actionable finding.
What is the broader impact of undiagnosed conditions on healthcare?
Diagnosis is not just a clinical endpoint. It is a systems-level pivot that reduces costs and enables targeted, effective care. Undiagnosed conditions increase health system costs through repeated investigations and emergency admissions, and a confirmed diagnosis enables more effective resource allocation across the entire care pathway. For health systems already under pressure, this is a compelling economic argument for investing in disease modeling infrastructure.
The public health implications reach further than individual cost savings. Undiagnosed patients in hospital settings carry what researchers call biological "dark matter," unidentified pathogens or novel disease presentations that standard surveillance misses entirely. Undiagnosed hospital patients are the frontline indicators of emerging pathogens, and health care systems that prioritize efficiency over exhaustive investigation create a critical blind spot for pandemic preparedness. Aligning diagnostic incentives with biosecurity goals is not an academic exercise. It is a practical necessity.
The contribution to precision medicine pipelines is equally significant. Rare disease models routinely illuminate mechanisms that later prove relevant to common diseases. The genetic pathways uncovered in rare metabolic disorders, for instance, have informed drug targets for type 2 diabetes and cardiovascular disease. This means the role of disease modeling in rare conditions generates scientific returns that extend across the entire disease spectrum.
Four systemic benefits of investing in undiagnosed disease models:
- Cost reduction: Confirmed diagnoses eliminate cycles of ineffective testing and reduce emergency admissions driven by unmanaged, unnamed conditions.
- Biosecurity: Systematic investigation of undiagnosed hospital patients detects novel pathogens before environmental surveillance does.
- Precision medicine: Rare disease mechanisms discovered through patient-derived models feed directly into broader drug development pipelines.
- Family and psychosocial support: Patient-centered multidisciplinary collaboration accelerates diagnosis and measurably reduces the emotional burden on families facing undiagnosed conditions.
"Diagnosis is both a scientific and a human challenge. When we solve an undiagnosed case, we are not just naming a disease. We are restoring a family's ability to plan, grieve, and move forward." — ERDERA Undiagnosed Day 2026
What are the main challenges in integrating these models into clinical practice?
The technical case for undiagnosed disease models is strong, but clinical integration remains uneven. Genomic data interpretation requires specialized bioinformatics expertise that most clinical centers do not have in-house. A whole-genome sequence generates millions of variants, and distinguishing pathogenic mutations from benign polymorphisms demands both computational tools and deep clinical genetics knowledge. Without that expertise, the data sits unused or, worse, gets misinterpreted.
Multidisciplinary collaboration is not optional in this field. It is the mechanism by which these models produce diagnoses. Geneticists, neurologists, biochemists, and patient advocates must work from shared data platforms to connect phenotypic observations with genomic findings. Programs like the NIH Undiagnosed Diseases Network succeed precisely because they pool expertise across institutions rather than relying on any single center's capabilities. The challenges in rare disease research are rarely purely scientific. They are organizational and structural.
Equity is the most underdiscussed barrier. Advanced genomic diagnostics and iPSC modeling are concentrated in academic medical centers in high-income countries. Patients in rural areas or lower-resource settings rarely access these tools, which means the diagnostic gap is not distributed evenly. Open data sharing platforms like ClinVar and Matchmaker Exchange partially address this by making variant interpretation data globally accessible, but the infrastructure to act on that data remains unequal.
Pro Tip: Clinicians managing suspected rare disease cases should submit unsolved genomic data to open sharing platforms like Matchmaker Exchange or the NIH Undiagnosed Diseases Network. These networks match phenotypic and genotypic data across institutions and have resolved cases that no single center could crack alone.
Emerging technologies are narrowing these gaps. Long-read sequencing platforms from companies like Pacific Biosciences and Oxford Nanopore Technologies now resolve structural variants and repeat expansions that short-read sequencing misses. Combined with AI-assisted variant prioritization tools, these advances are making faster rare disease answers accessible to a broader range of clinical settings. The trajectory is positive, but sustained funding and policy commitment are required to translate that trajectory into equitable access.
Key takeaways
Undiagnosed disease models are the most direct path from genomic ambiguity to clinical diagnosis, and their impact scales from individual patients to entire health systems.
| Point | Details |
|---|---|
| Diagnostic acceleration | Structured genomic programs reduce diagnostic timelines from 4 to 7 years down to 12 to 18 months. |
| Reanalysis value | Reanalyzing prior unsolved genomic data increases diagnostic yield by over 17%, converting cold cases into confirmed diagnoses. |
| Model superiority | Patient-derived iPSCs and organoids provide human-relevant 3D biology that animal models cannot replicate for rare genetic diseases. |
| Systemic cost impact | Confirmed diagnoses reduce repeated testing and emergency admissions, generating measurable savings for health systems. |
| Biosecurity relevance | Undiagnosed hospital patients are early indicators of novel pathogens, making disease modeling a pandemic preparedness tool. |
Why these models changed how I think about rare disease research
I have watched families spend years collecting binders of inconclusive test results, each specialist visit adding a new theory without resolution. What strikes me most about patient-derived disease models is not the technology itself. It is the shift in logic they represent. Traditional medicine asks, "What disease does this patient have?" Disease models ask, "What is this patient's biology actually doing?" That reframe changes everything.
The dual role of these models is what I find most underappreciated. They are simultaneously diagnostic tools and scientific discovery engines. An unsolved case that generates an iPSC line does not just help one patient. It becomes a research platform that can inform therapy development for every future patient with the same condition. The step-by-step genetic research process that produces these models is painstaking, but the compounding scientific return justifies every hour of it.
My honest concern is that funding and clinical adoption are not keeping pace with the science. The tools exist. The evidence is strong. What lags is the institutional will to build the multidisciplinary infrastructure these models require. Wider adoption will not happen through individual champions alone. It requires coordinated investment from health systems, governments, and biopharma partners who recognize that rare disease research is not a niche pursuit. It is a window into human biology that benefits everyone.
— John
How Hopeatrarelabs accelerates rare disease diagnosis and treatment
Hopeatrarelabs builds exactly the kind of patient-specific disease models this article describes, using iPSC technology and CRISPR editing to create functional platforms from patients' own cells. For researchers and clinicians who need to move from genomic data to testable biology without waiting years for academic pipelines, Hopeatrarelabs offers parallel treatment screens across FDA-approved drugs, custom antisense oligonucleotides, and gene therapy options.
The RareLabs Knowledge platform is a searchable resource built for patients, families, physicians, and biopharma partners who need centralized, evidence-based information on rare and undiagnosed conditions. Whether you are trying to understand a new diagnosis, identify treatment options, or connect with ongoing research, the knowledge base provides a structured starting point. Visit Hopeatrarelabs to explore how patient-derived modeling can move your case or research program forward.
FAQ
What are undiagnosed disease models?
Undiagnosed disease models are patient-specific biological platforms, including iPSC-derived cell lines, organoids, and CRISPR-edited tissue systems, built from a patient's own cells to study disease mechanisms and test potential therapies when no diagnosis or approved treatment exists.
How do disease models reduce diagnostic delays?
Structured genomic programs that incorporate functional disease modeling compress diagnostic timelines from an average of 4 to 7 years down to 12 to 18 months by providing direct functional evidence for candidate genetic variants rather than relying on clinical observation alone.
Why are patient-derived organoids better than animal models for rare diseases?
Patient-derived organoids replicate human-specific 3D tissue architecture and carry the patient's exact genetic variants, which animal models cannot reliably reproduce, making drug response data and phenotype-genotype correlations far more clinically relevant.
How do undiagnosed disease models support public health?
Undiagnosed hospital patients often carry the earliest signs of emerging pathogens. Systematic investigation of these cases through disease modeling provides biosecurity alerts that environmental surveillance alone misses, directly supporting pandemic preparedness.
Can previously unsolved genomic cases still be diagnosed?
Unsolved cases remain active research subjects. Reanalyzing previously negative genomic data with updated variant databases and new disease gene discoveries increases diagnostic yield by over 17%, meaning a case closed today may be solved within months or years as science advances.