
For many families dealing with ultra-rare or undiagnosed genetic diseases, the path to answers looks less like a straight line and more like a maze with no exit. Standard diagnostic tools miss the mark. Animal models built for common diseases don't capture the unique biology of a mutation that affects only a handful of people worldwide. Years pass, conditions worsen, and the question "what can we do?" stays unanswered. Genetic disease modeling recreates genetic disorders in controlled systems to study mechanisms, test therapies, and develop personalized treatments, offering a fundamentally different path forward for patients who have exhausted conventional options.
Table of Contents
- What is genetic disease modeling?
- The main technologies: iPSCs, organoids, and in silico models
- How genetic disease models drive diagnosis and personalized treatment
- Key limitations, ongoing challenges, and what's next
- Why every patient family should ask about personalized modeling
- Discover advanced genetic disease modeling solutions
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Personalized modeling power | Genetic disease modeling allows recreating a patient's disease in the lab, crucial for ultra-rare and undiagnosed cases. |
| Modern technologies explained | iPSCs, organoids, and in silico methods each play unique roles in understanding and treating rare disorders. |
| Real-world patient benefits | Lab-grown disease models speed up diagnosis and help find targeted treatments not possible with standard methods. |
| Know the challenges | No model is perfect—ask about validation, variability, and how labs keep up with rapidly evolving technology. |
| Take action | Families should actively seek out centers using these advanced personalized models for the best chance of progress. |
What is genetic disease modeling?
At its core, genetic disease modeling is the science of recreating a patient's disease in a controlled lab environment. Instead of guessing how a mutation behaves based on population averages, scientists use the patient's own cells or DNA to build a working replica of the disease. That replica becomes a testing ground where researchers can observe what's going wrong at the cellular level and experiment with potential fixes.
Here's why this matters so much for ultra-rare and undiagnosed conditions. If only 50 people in the world share your child's mutation, no pharmaceutical company has built a drug for it. No animal model has been specially engineered to carry it. No clinical trial exists. The conventional research pipeline simply doesn't serve you. Understanding genetic disease basics is the first step, but modeling takes that knowledge and translates it into actionable science.
The key advantages of patient-specific genetic disease modeling include:
- Capturing the exact mutation, not just a similar one used in standard research
- Reflecting the patient's genetic background, which influences how a disease actually presents
- Enabling drug screening on living human cells rather than animal tissue
- Bypassing the need for large patient cohorts that ultra-rare conditions simply can't provide
- Supporting functional testing, so researchers can see whether a potential treatment actually corrects the problem
"Genetic disease modeling is especially vital for ultra-rare and undiagnosed cases where no existing animal model captures the patient's specific disease mechanism."
Traditional animal models have real value for common diseases, but they fall short for patients whose conditions are defined by a single unique variant. A mouse engineered to carry a well-known BRCA mutation is useful for breast cancer research, but that same mouse tells you almost nothing about an uncharacterized variant in a gene affecting only a handful of known patients. The mismatch between the model and the disease produces unreliable results and wasted time that families simply don't have.
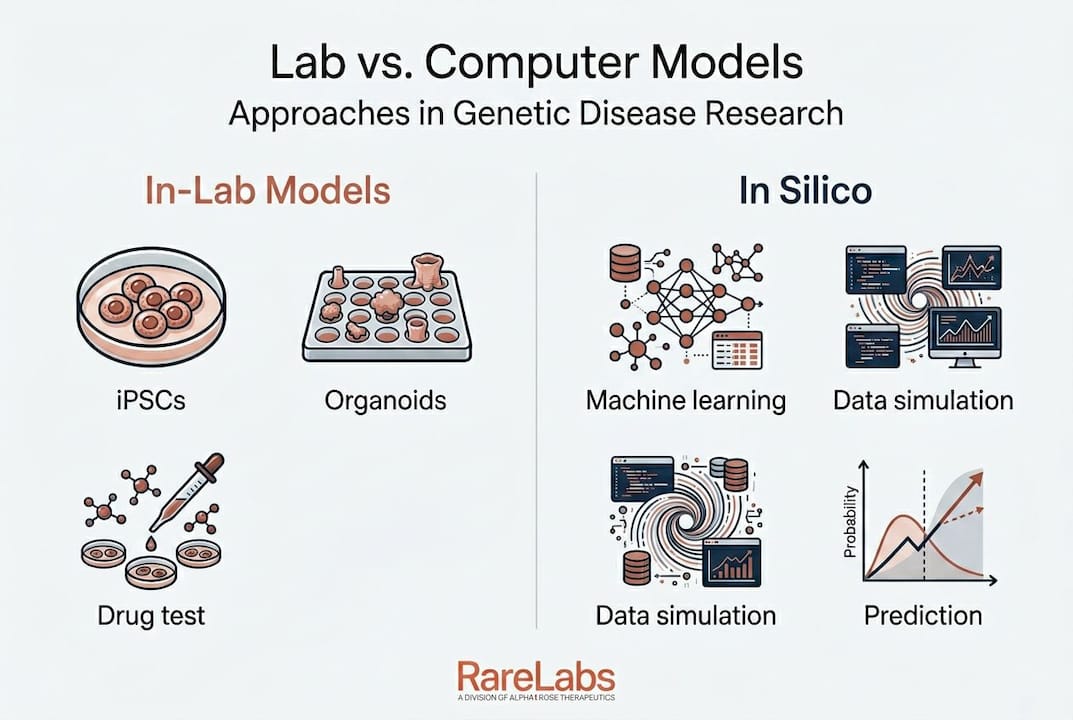
The main technologies: iPSCs, organoids, and in silico models
With the definition established, it's worth understanding the actual tools researchers use to build these models. Three platforms dominate the field right now, and each brings a unique set of strengths for ultra-rare disease work.

1. Induced pluripotent stem cells (iPSCs)
iPSCs are adult cells, often taken from a simple skin biopsy or blood draw, that are reprogrammed back into an embryonic-like state. From that state, scientists can guide them to become almost any cell type in the body: neurons, cardiomyocytes, hepatocytes, retinal cells, and more. iPSCs differentiated into 2D cells or 3D organoids retain the patient's full genetics, meaning every experiment conducted on those cells reflects what's actually happening in the patient's body. This is the foundation of truly personalized disease modeling.
2. Organoids
Organoids take iPSC technology one step further. Rather than growing cells in a flat two-dimensional layer, researchers coax them into three-dimensional mini-organ structures that self-organize to mimic real tissue architecture. A brain organoid captures spatial relationships between neurons. A retinal organoid reproduces the layered structure of the eye. This added complexity matters because many genetic diseases don't show their full effect in isolated cells. They emerge from the way cell populations interact in a tissue environment.
3. In silico models
In silico modeling uses artificial intelligence, machine learning, and computational simulations to predict disease behavior and drug responses. In silico methods predict drug responses and prevalence in virtual trials, which is especially useful for rare diseases where it's impossible to recruit large patient numbers for physical experiments. These tools can rank candidate gene variants by likelihood of pathogenicity and simulate how a drug molecule might interact with a mutant protein, all before a single cell is touched.
| Method | Key strength | Main limitation | Best use case |
|---|---|---|---|
| iPSC (2D) | Fast, scalable, cost-effective | Lacks tissue architecture | Initial drug screening |
| iPSC-organoids (3D) | High human fidelity, tissue context | Time-intensive, higher cost | Deep disease modeling |
| In silico / AI | Scalable, no cells needed | Requires validation, data scarce | Variant prioritization, virtual trials |
Pro Tip: If a lab offers only one of these platforms, ask whether combining iPSC-organoids with in silico tools is possible. The combination significantly improves personalization accuracy, especially for undiagnosed cases where the causal gene is still unknown.
Here's how a typical modeling workflow unfolds once patient cells arrive at the lab:
- Collect patient cells (blood, skin, or other accessible tissue)
- Reprogram cells into iPSCs using established protocols
- Differentiate iPSCs into the relevant disease-affected cell type
- Apply CRISPR editing to create isogenic controls (identical cells without the mutation) for direct comparison
- Conduct treatment screens using FDA-approved drugs, ASOs, or gene therapy vectors
- Validate results using in silico tools and compare against patient clinical data
How genetic disease models drive diagnosis and personalized treatment
Understanding platforms is useful, but what families really want to know is whether any of this translates to real answers and real treatments. It does, and the evidence is growing fast.
Organoids model monogenic disorders across brain, retina, and kidney, enabling gene discovery, drug screening, and CRISPR correction that would be impossible in animal models for ultra-rare variants. One of the clearest examples involves inherited retinal diseases like Stargardt disease. Retinal organoids derived from patient iPSCs reproduce the photoreceptor degeneration seen in patients, allowing researchers to test which interventions preserve cell function, something no mouse model fully replicates for rare ABCA4 variants.
For the truly undiagnosed, the combination of genomics and machine learning is producing measurable results. SHEPHERD ranks disease genes correctly in 40% of UDN cases, demonstrating that AI-driven genomic tools can meaningfully accelerate the gene discovery phase before physical modeling even begins. The Undiagnosed Diseases Network (UDN) handles some of the most complex cases in medicine, so a 40% gene-ranking accuracy in that population is a remarkable benchmark.
Here's a snapshot of recent breakthroughs using patient-specific disease models:
| Disease area | Modeling approach | Outcome |
|---|---|---|
| Stargardt disease (retinal) | Retinal organoids from patient iPSCs | Identified CRISPR correction targets |
| Undiagnosed neurological cases | Genomics + ML (SHEPHERD) | 40% correct gene ranking in UDN cases |
| Inherited kidney disorders | Kidney organoids from patient iPSCs | Functional rescue demonstrated post-correction |
| Rare cardiac arrhythmias | iPSC-derived cardiomyocytes | Drug response predictions validated clinically |
CRISPR-isogenic controls deserve special mention here. When scientists use CRISPR to create a cell line that is genetically identical to the patient's cells except for the corrected mutation, they get a direct comparison. Personalized rare disease therapy development moves significantly faster when the research doesn't have to account for background genetic noise. The signal, meaning the effect of the mutation, becomes much clearer.
For families exploring genetic therapy options, disease models also serve as an essential safety filter. Testing a gene therapy vector in a patient-derived organoid before clinical application gives researchers early warning signs about toxicity or off-target effects that would be invisible in a standard cell line.
Key clinical applications of disease models currently in active use:
- Parallel drug screens: Testing hundreds or thousands of FDA-approved compounds simultaneously against patient-derived cells
- ASO design validation: Confirming that custom antisense oligonucleotides correct splicing or expression defects in patient cells
- Gene therapy vector testing: Evaluating delivery efficiency and safety in relevant cell types before any clinical application
- Biomarker discovery: Identifying measurable indicators of disease progression or treatment response unique to the patient's mutation
Key limitations, ongoing challenges, and what's next
Honest science requires acknowledging what these tools cannot yet do. Genetic disease modeling is powerful, but it isn't perfect, and families deserve a clear-eyed understanding of its current boundaries.
Variability in iPSC lines and incomplete pathology recapitulation remain real technical challenges. Late-onset conditions are particularly difficult: a disease that takes 40 years to damage neurons in a person may not reproduce that timeline in a lab model that exists for weeks or months. Complex interactions between multiple genetic variants, environmental factors, and aging are hard to capture fully. Researchers address this through rigorous line selection and multiple independent experiments, but variability is something any honest lab will acknowledge.
Scaling is another issue. In silico models are scalable but limited by data scarcity and risk overfitting when trained on small, non-diverse datasets. Ultra-rare diseases, by definition, produce small datasets. This creates a circular problem: the rarer the disease, the more you need AI help, but the less training data exists to make that AI reliable. Hybrid approaches that combine wet-lab validation with computational predictions are the most practical current solution.
Ethical considerations also shape what modeling labs can and should do. Patient-derived cells carry sensitive genetic information. Data sharing between institutions could accelerate rare disease research enormously, but it requires careful consent frameworks and privacy protections that don't yet exist uniformly across the field.
Addressing key rare disease challenges in modeling requires the whole ecosystem to improve. The most promising near-term directions include:
- CRISPR-organoid hybrid platforms that accelerate functional correction studies
- Multi-omics integration combining genomics, proteomics, and metabolomics data from the same patient model
- Bioengineered scaffolds that give organoids more physiologically relevant structural support
- Federated AI training across multiple rare disease centers to build larger, more diverse datasets without compromising patient privacy
Pro Tip: When evaluating a modeling lab for your disease, ask specifically how they manage iPSC line variability, how many independent lines they use, and what validation steps they take before reporting results. A rigorous lab will have clear, documented answers.
Why every patient family should ask about personalized modeling
Here's something that doesn't get said enough: most families navigating ultra-rare or undiagnosed diagnoses never hear about patient-specific modeling as an option. They're told the disease is too rare for research, that no model exists, or that the only path is a compassionate use request for a drug designed for a different condition entirely. That framing is outdated.
The technology exists right now. iPSC-organoid platforms are not experimental fantasies waiting for the next decade of development. They are operating labs conducting treatment screens on patient cells today. The gap isn't technological capability. It's awareness and access.
What families often encounter is a system that defaults to population-level solutions for diseases that are, by their nature, individual. A drug that works for the most common mutation in a given gene might do nothing, or even cause harm, for a patient carrying a private variant in that same gene. Personalized modeling is precisely what addresses this mismatch.
The families who move fastest are those who stop waiting for the medical system to offer this and start seeking labs that specialize in it. Asking your neurologist about iPSC modeling may get a blank stare. But reaching out directly to a specialized biotech lab with experience in must-know gene therapy approaches and patient-derived modeling gets you into a completely different conversation.
The future of rare disease treatment is personal, and that future is accessible now for families willing to seek it out. Hybrid AI-driven platforms, multi-omics integration, and CRISPR-isogenic controls are not science fiction. They are tools available at specialized labs today. The question isn't whether this technology works. It's whether your family knows to ask for it.
Discover advanced genetic disease modeling solutions
If this article has shifted how you think about your family's options, the next step is finding the right resources and lab partners to act on that knowledge. Families navigating ultra-rare or undiagnosed conditions often feel like they're searching in the dark. Specialized knowledge changes that equation immediately.
At RareLabs, we build patient-specific models using iPSCs, organoids, and CRISPR gene editing to test thousands of FDA-approved drugs, custom ASOs, and gene therapy options against your disease, not a population average. Explore our rare disease knowledge hub to understand the latest tools available and the science behind personalized treatment screens. When you're ready to take the next step, visit RareLabs to learn how a personalized rare disease search strategy can accelerate your path from uncertainty to answers.
Frequently asked questions
How are organoids different from animal models in genetic disease research?
Organoids are miniature lab-grown versions of human organs made from patient cells, allowing for personalized modeling and higher human fidelity. Organoids and iPSCs are superior for human fidelity and personalization compared to animal models, which often fail to capture unique patient-specific variants.
What makes genetic disease modeling important for undiagnosed or ultra-rare conditions?
It enables personalized study of the patient's own cells, allowing researchers to identify the disease mechanism and test custom treatment options when standard tools fail. Genetic disease modeling is especially vital for ultra-rare and undiagnosed cases that lack existing animal models or approved therapies.
How accurate are genetic disease models for predicting real-life drug responses?
iPSC-organoids can closely mimic patient biology and show measurable functional rescue after treatment, but results must always be validated before clinical application. ML classifiers achieve AUC 0.86 for disease severity prediction, reflecting strong but imperfect performance that still requires wet-lab confirmation.
What questions should families ask labs offering modeling for their disease?
Ask about the model types used, how they manage iPSC line variability, what validation steps are applied, and whether they integrate AI or hybrid approaches. Variability in iPSC lines and incomplete recapitulation are real concerns, and any credible lab will have transparent protocols for addressing them.