
Rapid therapy discovery is the process of compressing drug development timelines from a decade or more down to months, and for patients with rare and undiagnosed genetic diseases, that compression is the difference between a treatment and a death sentence. Why rapid therapy discovery matters becomes undeniable when you consider that traditional drug development spans 10 to 12 years, while most rare disease patients cannot wait that long. AI systems like Robin, FDA programs like the National Priority Voucher (CNPV), and patient-derived modeling platforms are now rewriting what is biologically and regulatorily possible. Hopeatrarelabs operates at the center of this shift, building personalized disease models from patients' own cells to run parallel treatment screens that would have been unthinkable five years ago.
Why rapid therapy discovery matters in AI-enabled drug development
Artificial intelligence has become the most significant accelerator in early-stage drug discovery, and the numbers behind that claim are not incremental. BCG and Wellcome Trust analysis projects that AI can reduce discovery and preclinical stage time and cost by 25 to 50%. That means a phase that once consumed three to four years could realistically complete in 18 months, freeing resources and attention for the clinical work that follows.
The Robin AI system illustrates what this looks like in practice. Robin analyzed 551 papers in 30 minutes, a task that would require an estimated 540 hours of manual processing by a research team. The implication is not just speed. It is the ability to surface target candidates that a human team would statistically miss, particularly for ultra-rare conditions where published literature is sparse and scattered across disciplines.

Understanding rapid therapy development through an AI lens requires honesty about what AI cannot do. Machine learning models excel at pattern recognition across large datasets, but they cannot perform biological causal inference with the same reliability. A model can predict that a compound binds to a target. It cannot yet reliably predict whether that binding produces a therapeutic effect in a living system with a specific genetic mutation. Human-led experimental validation remains non-negotiable.
The practical architecture that works combines AI-generated candidate lists with wet-lab confirmation using patient-derived models. Induced pluripotent stem cells (iPSCs) reprogrammed from a patient's own cells give researchers a biologically accurate substrate to test AI-nominated compounds. This pairing is where the real acceleration happens, not in AI alone.
- AI reduces literature review from hundreds of hours to minutes, enabling faster target identification
- iPSC-based models provide patient-specific validation that generic cell lines cannot replicate
- Parallel screening of FDA-approved drugs, antisense oligonucleotides (ASOs), and gene therapy vectors compresses candidate selection
- Human researchers remain responsible for interpreting biological outcomes and advancing candidates to clinical stages
Pro Tip: When evaluating AI-generated drug candidates for rare genetic diseases, prioritize platforms that integrate patient-derived iPSC validation in the same workflow. Candidates confirmed in patient-specific models carry significantly higher translational credibility than those validated only in standard cell lines.
How FDA real-time trial programs and priority vouchers speed approvals
Regulatory timelines have historically been as much of a bottleneck as scientific discovery. The standard review window runs 10 to 12 months after a Biologics License Application or New Drug Application filing. For a patient with a progressive genetic disease, that window is clinically meaningful in the worst possible way.
The FDA's National Priority Voucher (CNPV) pilot changes that equation. The fastest CNPV approval on record was 61 days post-BLA filing. That is not a rounding error on the standard timeline. It is a structural redesign of how regulatory review operates, and it required early Chemistry, Manufacturing, and Controls (CMC) readiness as a precondition.
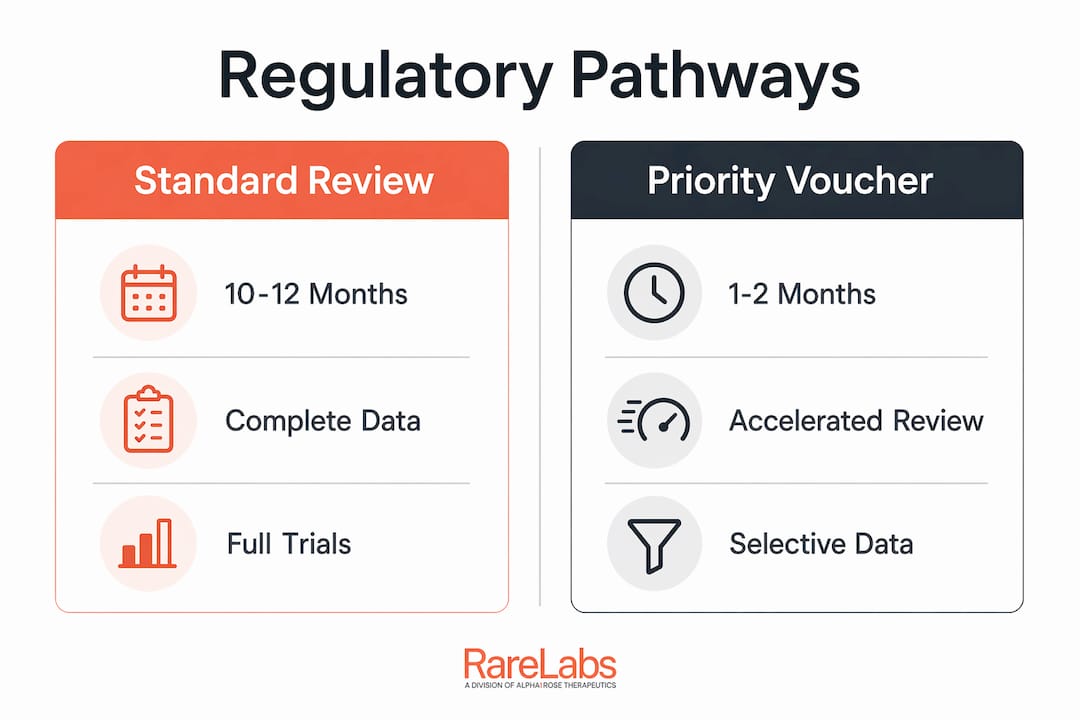
The FDA's real-time clinical trial pilots take a parallel approach to the trial phase itself. Rather than submitting data in batches at the end of a trial, sponsors submit data continuously as it is generated. Reviewers assess it in real time, which means the agency is not starting its review from zero when the trial ends. The practical effect is that database lock to market entry can shrink from years to months.
Here is what operational readiness for these programs actually requires:
- CMC documentation must be submission-ready before the trial ends, not assembled afterward
- Regulatory affairs teams must be embedded from Phase 2 onward, not brought in at filing
- Data management infrastructure must support continuous submission rather than batch export
- Manufacturing scale-up must be aligned with approval timelines to prevent post-approval supply gaps
- Talent with experience in real-time review programs is scarce and must be recruited early
The data table below summarizes the timeline contrast between standard and accelerated regulatory pathways.
| Pathway | Review duration | Key requirement |
|---|---|---|
| Standard FDA review | 10 to 12 months | Complete BLA/NDA submission |
| CNPV pilot | 1 to 2 months | Early CMC readiness, priority designation |
| Real-time trial program | Concurrent with trial | Continuous data submission infrastructure |
Pro Tip: If your organization is targeting a CNPV designation, treat CMC readiness as a Phase 2 deliverable, not a Phase 3 task. Sponsors who arrive at filing with incomplete manufacturing documentation forfeit the timeline advantage entirely.
What biological and operational realities actually limit discovery speed
The significance of fast treatment development is real, but so are the constraints that no technology or regulation can eliminate. Expert René Kuijten frames this precisely:
"Biology has irreducible time costs. You cannot compress a cell differentiation cycle or a patient's immune response the way you can compress a literature review. Acceleration means removing artificial delays, not skipping validation."
This distinction matters enormously for researchers and clinicians who communicate timelines to patients and families. AI and regulatory reform remove administrative and analytical delays. They do not change the time required for a cell to differentiate, a biomarker to express, or a clinical cohort to accumulate sufficient follow-up data. Biological validation carries time costs that are genuinely irreducible.
The funding structure compounds this problem. The 5 to 7 year translation valley between basic discovery and late-stage clinical trials is chronically underfunded. Most promising science fails here, not because it lacks merit, but because it lacks the patient capital required to sustain multi-year development without near-term commercial returns. Rare disease programs are especially vulnerable because the addressable market is small and traditional venture timelines do not align with biological reality.
Operational bottlenecks add a third layer. Patient recruitment and global trial coordination are consistently the rate-limiting steps in rare disease trials, not scientific discovery. A trial for a condition affecting 200 patients worldwide cannot be accelerated by better algorithms. It requires patient registries, international site coordination, and the kind of infrastructure that takes years to build. Researchers who conflate scientific acceleration with operational acceleration consistently underestimate how long trials actually take.
The honest framing for healthcare professionals is this: AI and regulatory reform can realistically compress the front and back ends of the development pipeline. The middle, where biology, funding, and operations intersect, remains the hardest and most consequential phase to accelerate.
Collaborative strategies that actually accelerate rare disease therapy delivery
The advantages of accelerated therapy research become concrete when organizations build the right collaborative architecture rather than relying on any single innovation. The most effective approaches combine patient-derived biology, cross-sector partnerships, and early regulatory integration.
Patient-derived iPSC models are the foundation. When a research team screens compounds against a model built from the patient's own cells, they generate data that is directly translatable to that individual's disease mechanism. This is not a marginal improvement over standard cell lines. It is a qualitatively different category of evidence, and it is why personalized research consistently outperforms population-average approaches for rare genetic conditions.
Partnerships between pharma organizations and AI labs accelerate knowledge transfer in both directions. Legacy pharma brings clinical development infrastructure, regulatory relationships, and manufacturing capacity. AI labs bring target identification speed, compound library analysis, and data integration capabilities. Neither has the full stack alone. The organizations moving fastest in rare disease therapy development in 2026 are those that have formalized these partnerships rather than treating them as experimental collaborations.
Embedding regulatory affairs from Phase 2 onward is not a bureaucratic preference. It is a timeline decision. Teams that involve regulatory strategists early identify CMC gaps, labeling issues, and data package requirements before they become filing delays. The drug repurposing pathway offers a specific example where early regulatory alignment dramatically shortens approval timelines because the safety profile of an FDA-approved compound is already established.
- Build patient registries before trials begin, not during recruitment
- Use parallel treatment screens to test multiple compound classes simultaneously rather than sequentially
- Align manufacturing scale-up with Phase 3 timelines to prevent post-approval supply delays
- Maintain data transparency with patients and families throughout the process to support retention and trust
The peptide biomarker research emerging from specialized labs is also reshaping how researchers validate targets earlier in the process, reducing the risk of late-stage failures that waste years of development time.
Key takeaways
Rapid therapy discovery compresses development timelines by removing artificial delays through AI, regulatory reform, and collaborative infrastructure, while biological validation time costs remain irreducible.
| Point | Details |
|---|---|
| AI cuts early-phase timelines | BCG and Wellcome Trust project 25 to 50% reductions in discovery and preclinical phases via AI. |
| CNPV pilot approval in 61 days | FDA's National Priority Voucher reduced review from 10 to 12 months down to as few as 61 days with early CMC readiness. |
| Biology has irreducible time costs | AI and regulation remove administrative delays but cannot compress cell differentiation cycles or clinical follow-up requirements. |
| Translation valley is underfunded | The 5 to 7 year gap between discovery and late-stage trials fails most promising science due to lack of patient capital. |
| Collaboration is the operational key | iPSC models, pharma-AI partnerships, and early regulatory integration are the practical levers that accelerate rare disease therapy delivery. |
The uncomfortable truth about acceleration timelines
I have spent years watching organizations announce AI-powered drug discovery platforms with timelines that would make a biologist wince. The enthusiasm is understandable. The 25 to 50% reduction in early-phase timelines that BCG and Wellcome Trust project is real and significant. But the gap between what AI can do at the discovery stage and what the full development pipeline actually requires is where most of the overclaiming happens.
What I have found actually works is a more modest framing: AI removes the delays we created, not the delays biology requires. Robin analyzing 551 papers in 30 minutes is genuinely transformative for target identification. It does not change how long it takes to confirm that a target is therapeutically relevant in a patient-derived model. Those are different problems.
The part of this conversation that rarely gets enough attention is the translation valley. Sustained NIH funding and patient capital structures that match biological timelines are not glamorous topics. They do not generate press releases. But the 5 to 7 year underfunded middle phase is where more promising rare disease science dies than at any other point in the pipeline. Fixing that requires policy commitment and institutional patience, not better algorithms.
My honest view is that the rare disease field is at an inflection point that is real but fragile. The regulatory innovations at FDA, the maturation of iPSC modeling, and the genuine capabilities of AI target identification have created a window where timelines can compress meaningfully. Sustaining that requires building the talent ecosystems, funding structures, and operational infrastructure that make acceleration durable rather than episodic. Patients with rare genetic diseases deserve both the urgency and the honesty.
— John
How Hopeatrarelabs accelerates your rare disease therapy search
Hopeatrarelabs was built specifically for the gap this article describes: the space between a diagnosis with no approved treatment and a therapy that might work for that specific patient.
The RareLabs Knowledge hub gives healthcare professionals, researchers, and patient advocates direct access to the latest rare disease research, treatment screening results, and therapy options across FDA-approved drugs, ASOs, and gene therapy candidates. Every resource is organized around the urgency that rare disease cases demand. If you are working on a case with no clear treatment path, the rare disease treatment search platform lets you search across experimental and approved options using patient-specific parameters. The tools exist. The question is whether you are using them.
FAQ
What is rapid therapy discovery?
Rapid therapy discovery refers to the use of AI, patient-derived disease models, and regulatory innovations to compress drug development timelines from 10 to 12 years toward months for specific disease targets. It is most critical for rare and undiagnosed genetic diseases where no approved treatments exist.
How does AI reduce drug development timelines?
AI systems like Robin reduce target identification time by analyzing thousands of research papers in minutes rather than hundreds of hours. BCG and Wellcome Trust project that AI can cut discovery and preclinical phase time and cost by 25 to 50%.
What is the FDA's National Priority Voucher program?
The CNPV pilot reduces FDA review time from the standard 10 to 12 months down to 1 to 2 months for qualifying drugs. The fastest approval on record was 61 days post-BLA filing, contingent on early CMC readiness.
Why can't biology be fully accelerated?
Biological processes like cell differentiation, immune response, and clinical follow-up have irreducible time requirements that AI and regulatory reform cannot compress. Acceleration removes administrative and analytical delays, not the time biology itself requires for validation.
What is the translation valley in drug development?
The translation valley is the 5 to 7 year underfunded phase between basic scientific discovery and late-stage clinical trials. Most promising rare disease science fails here due to lack of patient capital and misaligned funding timelines, not scientific inadequacy.