
Протеостаз — это система клеточного контроля качества белков, нарушение которой лежит в основе патогенеза большинства редких генетических заболеваний. Клетка синтезирует, сворачивает, транспортирует и разрушает тысячи белков одновременно. Когда этот баланс нарушается из-за генетической мутации, дефектные белки накапливаются и разрушают ткань изнутри. Понимание роли протеостаза при редких генетических болезнях открывает путь к терапии, которая работает не с симптомами, а с молекулярной причиной болезни. Именно здесь сосредоточены наиболее перспективные исследования 2026 года.
Как нарушения протеостаза вызывают редкие генетические болезни?
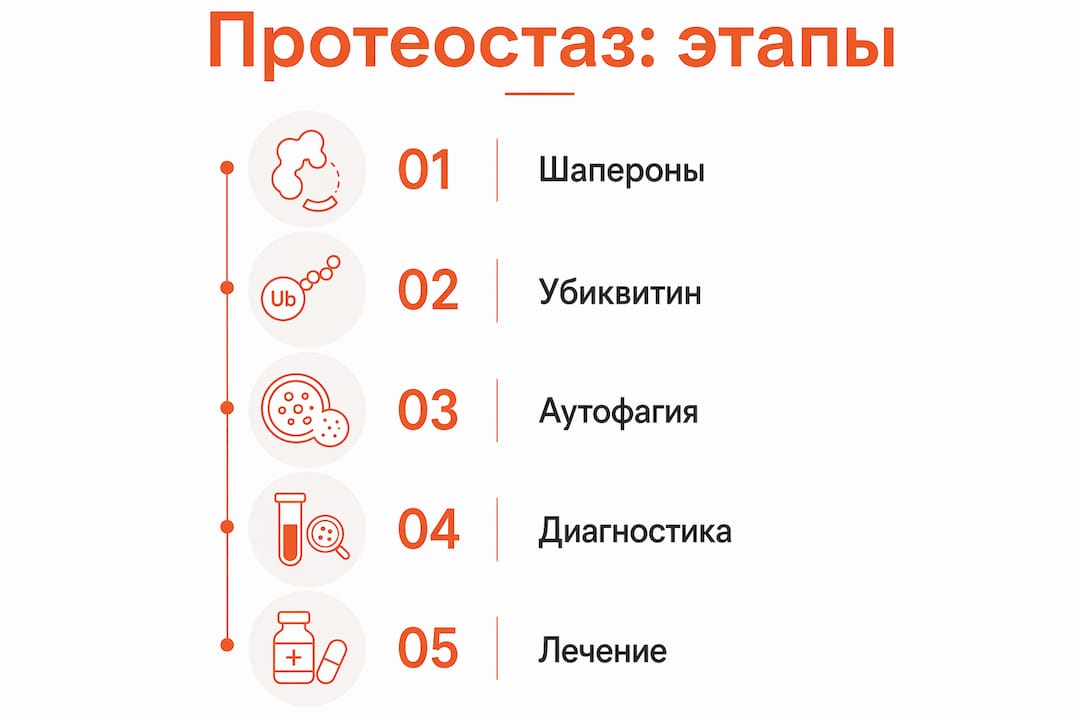
Протеостаз держится на трёх опорах: шапероны складывают белки правильно, убиквитин-протеасомная система уничтожает дефектные молекулы, аутофагия убирает крупные агрегаты. Сбой в любом из этих звеньев ведёт к накоплению токсичных белков. Нарушение протеостаза связано с накоплением амилоид-бета, тау-белка и альфа-синуклеина при нейродегенеративных болезнях, а при наследственных заболеваниях механизм тот же, только запускается генетической мутацией с рождения.
Генетические нарушения и протеостаз связаны напрямую: мутация меняет структуру белка так, что шапероны не могут его правильно свернуть. Белок либо теряет функцию, либо приобретает токсичные свойства. Профессор А. Белогуров описывает это как «запирание» болезни в клетке через ошибки синтеза белков. Это означает, что без вмешательства в протеостаз генная терапия сама по себе не всегда достаточна.

Конкретные примеры показывают масштаб проблемы. При наследственных подоцитопатиях у 79,2% детей с нефротическим синдромом выявлены моногенные мутации, нарушающие структурные белки подоцитов. Это разрушает фильтрационную функцию почек через механизм протеостатической перегрузки. При дисплазии соединительной ткани патогенные варианты в генах COL3A1 и FGFR3 вызывают структурные дефекты белков коллагена и рецепторов роста. Ткань теряет механическую прочность, потому что дефектные белки не разрушаются вовремя.
Ключевые механизмы нарушения протеостаза при редких генетических болезнях:
- Перегрузка шаперонов. Мутантные белки требуют больше ресурсов для сворачивания. Шапероны отвлекаются от нормальных белков, и клетка теряет функциональность.
- Дисфункция протеасомы. Дефектные белки блокируют протеасому, снижая её пропускную способность. Это создаёт цепную реакцию накопления токсичных молекул.
- Нарушение аутофагии. При некоторых мутациях аутофагосомы не формируются или не сливаются с лизосомами. Крупные белковые агрегаты остаются в клетке.
- Стресс эндоплазматического ретикулума (ЭПР). Накопление несвёрнутых белков в ЭПР запускает UPR-ответ. При хроническом стрессе UPR переключается с защиты на апоптоз.
Профессиональный совет: При анализе клинических случаев редких генетических болезней всегда проверяйте, какое именно звено протеостаза затронуто мутацией. Это определяет выбор терапевтической мишени точнее, чем фенотипическое описание.
Какие методы диагностики позволяют оценить протеостаз при редких болезнях?
Диагностика нарушений протеостаза требует нескольких уровней анализа одновременно. Генетический анализ выявляет мутацию, но не показывает, как именно она влияет на белковый баланс в конкретной ткани. Поэтому современная диагностика сочетает геномные, протеомные и клеточные методы.
-
Высокопроизводительное секвенирование (ВПС). Метод выявляет мутации в генах, кодирующих компоненты протеостатической сети. При дисплазии соединительной ткани ВПС позволил идентифицировать мутантные гены у 14 пациентов с разной клинической выраженностью. Это показывает, что один и тот же ген может давать разные фенотипы в зависимости от контекста протеостаза.
-
Протеомика. Масс-спектрометрия выявляет изменения в составе белкового пула клетки. Она показывает, какие белки накапливаются, а какие деградируют быстрее нормы. Это прямая карта нарушения протеостаза.
-
Транскриптомика. Роль транскриптомики в моделировании редких болезней состоит в том, что она показывает, какие гены шаперонов и убиквитин-лигаз активированы или подавлены. Это позволяет предсказать, на каком этапе протеостаз даёт сбой ещё до появления клинических симптомов.
-
Модели на основе iPSC. Индуцированные плюрипотентные стволовые клетки, полученные от конкретного пациента, воспроизводят его уникальное нарушение протеостаза в лаборатории. Персонализированное тестирование на iPSC позволяет проверить тысячи молекул для восстановления протеостаза у пациентов с ультра-редкими мутациями. Это часто единственный способ найти ответ, когда мутация встречается у единиц людей в мире.
-
Функциональные клеточные анализы. Флуоресцентные репортёры и анализы агрегации белков в живых клетках показывают динамику протеостаза в реальном времени. Они дополняют геномные данные функциональным контекстом.
Профессиональный совет: Не ограничивайтесь ВПС при диагностике редких болезней. Протеомный анализ ткани-мишени часто выявляет нарушения протеостаза, которые геномный анализ пропускает, особенно при вариантах неизвестного значения.
Как протеостаз становится мишенью для новых методов лечения?
Терапия, нацеленная на сеть протеостаза, может иметь преимущество над прямой генетической заменой. Это особенно важно при болезнях, где мутация не устраняется, но её последствия для белкового баланса можно скорректировать фармакологически.
Механизм действия артемизинина стал модельным примером такого подхода. Артемизинин ингибирует протеасому паразита, вызывая накопление повреждённых белков и гибель клетки-мишени. Тот же принцип, применённый в обратную сторону, активация протеасомы или шаперонов, может спасать клетки пациента от токсичных агрегатов.
Фармакологическая модуляция стресса эндоплазматического ретикулума и баланс аутофагии — ключ к успешной терапии, но требует точного контроля. Чрезмерная активация аутофагии может повреждать клетки. Поиск терапевтического окна — главный вызов для исследователей в 2026 году.
Основные терапевтические стратегии, направленные на протеостаз:
- Фармакологические шапероны. Малые молекулы стабилизируют мутантный белок в правильной конформации. Этот подход уже применяется при болезни Гоше и некоторых формах муковисцидоза.
- Ингибиторы и активаторы протеасомы. Регуляция скорости деградации белков позволяет убирать токсичные агрегаты или, напротив, сохранять нестабильные, но функциональные белки.
- Модуляция аутофагии. Рапамицин и его аналоги активируют аутофагию через ингибирование mTOR. Это ускоряет клиренс белковых агрегатов при лизосомных болезнях накопления.
- Антисмысловые олигонуклеотиды (ASO). ASO корректируют сплайсинг или снижают синтез токсичного белка на уровне мРНК. Hopeatrarelabs разрабатывает персонализированные ASO для пациентов с ультра-редкими мутациями.
- Генная терапия в сочетании с протеостатической поддержкой. Доставка правильного гена эффективнее, если одновременно снизить нагрузку на протеостаз клетки-хозяина.
Примеры успешного лечения редких заболеваний в 2026 году показывают: комбинированные стратегии, сочетающие генную терапию с модуляцией протеостаза, дают лучшие результаты, чем каждый подход по отдельности. Это меняет логику разработки терапии для редких болезней.
Что важно знать семьям и исследователям о протеостазе при редких болезнях?
Тканеспецифичность протеостаза — один из наименее очевидных, но критически важных аспектов. Локальное истощение системы приводит к гибели специфических клеток, даже если протеостаз в других органах работает нормально. Именно поэтому одна и та же мутация может разрушать почки, но не затрагивать печень, или поражать нейроны, оставляя мышечную ткань интактной.
Для семей пациентов это означает несколько практических выводов:
- Раннее вмешательство критично. Ранняя терапия замедляет необратимое накопление белковых агрегатов. После того как агрегаты сформировались, их устранение требует значительно больших усилий и не всегда возможно.
- Мониторинг должен быть тканеспецифичным. Общие биомаркеры воспаления или функции органов не отражают состояние протеостаза в клетке-мишени. Нужны специфические маркеры для каждого типа ткани.
- Генетический диагноз — это начало, а не конец. Знание мутации не объясняет, как именно нарушен протеостаз у конкретного пациента. Функциональный анализ на iPSC-моделях даёт этот ответ.
- Психологическая поддержка семей необходима. Ожидание результатов молекулярного анализа и неопределённость прогноза создают значительную нагрузку. Семьи нуждаются в сопровождении специалистов, понимающих специфику редких болезней.
Профессиональный совет: Если ваш пациент или близкий получил диагноз редкого генетического заболевания, запросите функциональный анализ белкового баланса в ткани-мишени. Это позволит выбрать терапевтическую мишень точнее, чем только на основании генетического варианта.
Персонализированная медицина на основе iPSC меняет подход к редким болезням. Клетки пациента перепрограммируются в нужный тип ткани, и исследователи видят нарушение протеостаза именно там, где оно происходит. Это устраняет разрыв между геномным диагнозом и реальной клеточной патологией. Типы редких генетических заболеваний крайне разнообразны, и iPSC-модели позволяют работать с каждым случаем индивидуально, а не по общему протоколу.
Ключевые выводы
Роль протеостаза при редких генетических болезнях определяет не только патогенез, но и терапевтическую стратегию: восстановление белкового баланса в клетке-мишени эффективнее, чем симптоматическое лечение.
| Пункт | Подробности |
|---|---|
| Три опоры протеостаза | Шапероны, убиквитин-протеасомная система и аутофагия работают вместе; сбой в одном звене разрушает всю систему. |
| Тканеспецифичность | Одна мутация поражает конкретные ткани, потому что локальная белковая нагрузка в них выше, чем в других органах. |
| Раннее вмешательство | Терапия до формирования белковых агрегатов предотвращает необратимые повреждения клеток. |
| iPSC как инструмент диагностики | Модели на основе клеток пациента показывают реальное нарушение протеостаза в ткани-мишени. |
| Фармакологическая модуляция | Шапероны, регуляторы аутофагии и ASO корректируют протеостаз без замены гена. |
Протеостаз: почему мы недооцениваем главное
За годы работы с редкими генетическими болезнями я пришёл к одному неудобному выводу: большинство терапевтических неудач происходит не потому, что мы не знаем мутацию. Мы её знаем. Неудачи случаются потому, что мы игнорируем состояние протеостаза в ткани-мишени на момент начала лечения.
Генная терапия доставляет правильную копию гена в клетку, которая уже перегружена токсичными агрегатами. Новый белок синтезируется, но шапероны заняты, протеасома перегружена, и правильный белок просто не успевает сложиться. Терапия технически работает, но клинического эффекта нет. Это не гипотеза. Это паттерн, который повторяется в клинических данных снова и снова.
Второй момент, который меня поражает: исследователи часто рассматривают протеостаз как фоновый процесс, а не как активную терапевтическую мишень. Между тем фармакологические шапероны при болезни Гоше показали, что можно восстановить функцию мутантного белка без его замены. Это переворачивает логику разработки терапии.
Семьям пациентов я говорю прямо: требуйте от врачей не только генетического диагноза, но и функционального анализа. Спрашивайте, какое звено протеостаза нарушено. Это вопрос, который меняет выбор лечения. Будущее терапии редких болезней строится на сочетании генетической точности и протеостатической поддержки. Одно без другого работает хуже.
— John
Hopeatrarelabs: персонализированный подход к протеостазу при редких болезнях
Hopeatrarelabs создаёт персонализированные модели болезней на основе клеток конкретного пациента, используя технологии iPSC и редактирование генома CRISPR. Это позволяет воспроизвести нарушение протеостаза в ткани-мишени и протестировать тысячи молекул, включая одобренные FDA препараты и персонализированные ASO, для восстановления белкового баланса.
Платформа RareLabs Knowledge Resource объединяет инструменты для работы с редкими генетическими заболеваниями: от молекулярного моделирования до анализа терапевтических опций. Для семей, врачей и исследователей, которые ищут ответ при ультра-редких мутациях, Hopeatrarelabs предлагает прозрачный и научно обоснованный процесс поиска терапии. Узнайте больше о возможностях платформы и начните персонализированный поиск терапии уже сегодня.
Часто задаваемые вопросы
Что такое протеостаз и почему он важен при редких болезнях?
Протеостаз — это система клеточного контроля качества белков, включающая шапероны, протеасому и аутофагию. При редких генетических болезнях мутации нарушают этот баланс, что приводит к накоплению токсичных белков и гибели клеток.
Какие гены чаще всего нарушают протеостаз при редких заболеваниях?
Мутации в генах NPHS2, COL3A1 и FGFR3 — типичные примеры. Они изменяют структуру белков так, что шапероны не могут их правильно свернуть, а протеасома не успевает их разрушить.
Как iPSC-модели помогают в изучении протеостаза?
iPSC-модели воспроизводят нарушение протеостаза в клетках конкретного пациента. Это позволяет тестировать терапевтические молекулы в условиях, точно соответствующих реальной патологии, что особенно ценно при ультра-редких мутациях.
Можно ли восстановить протеостаз без генной терапии?
Да. Фармакологические шапероны, модуляторы аутофагии и антисмысловые олигонуклеотиды корректируют белковый баланс без замены гена. При болезни Гоше этот подход уже применяется клинически.
Почему раннее вмешательство при нарушениях протеостаза так важно?
Белковые агрегаты, накопившиеся в клетке, крайне сложно устранить. Ранняя терапия предотвращает их формирование и сохраняет функцию клетки, тогда как поздняя терапия работает с уже необратимыми повреждениями.