
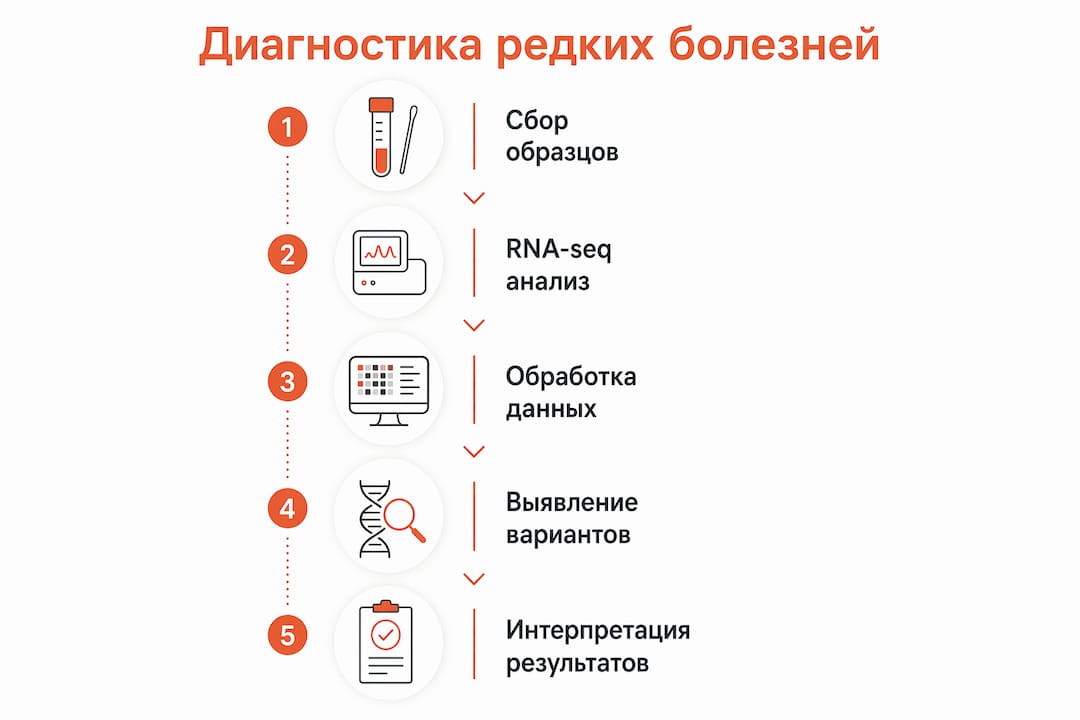
Транскриптомика определяется как анализ всей совокупности РНК-молекул клетки для изучения активности генов в конкретный момент времени. Именно этот метод становится ключевым инструментом в моделировании редких генетических заболеваний: он позволяет перейти от статичного описания мутаций к функциональному пониманию того, как они нарушают работу клетки. До 40% пациентов с подозрением на моногенные заболевания не получают окончательного диагноза после стандартного секвенирования ДНК. Роль транскриптомики в моделировании редких болезней состоит в том, чтобы заполнить этот диагностический пробел через функциональную интерпретацию генетических вариантов и построение точных клеточных моделей.
Какие технологии транскриптомики используются для моделирования редких генетических заболеваний?
Пространственная и одноклеточная транскриптомика меняют традиционный взгляд на архитектуру болезни. Если стандартное секвенирование даёт усреднённый сигнал по всей ткани, то одноклеточная транскриптомика (scRNA-seq) разрешает профиль экспрессии на уровне каждой отдельной клетки. Пространственная транскриптомика добавляет координаты: исследователь видит не только какие гены активны, но и где именно в ткани это происходит. Для редких патологий с гетерогенной клеточной структурой это критически важно.
Технологии, применяемые сегодня для построения транскриптомных моделей редких заболеваний:
- scRNA-seq (одноклеточная РНК-секвенирование): выявляет редкие клеточные субпопуляции, которые теряются при анализе смешанных образцов.
- Пространственная транскриптомика (Visium, Slide-seq): сохраняет топографию экспрессии внутри ткани, что позволяет изучать микросреду патологического очага.
- Perturb-seq: объединяет CRISPR-скрининг с транскриптомным профилированием. Каждая клетка получает определённое генетическое возмущение, а её транскриптомный ответ фиксируется одновременно.
- Bulk RNA-seq: остаётся стандартом для первичного скрининга экспрессии при ограниченном материале.
- Мультиомиксная интеграция: объединяет транскриптомные данные с протеомикой, метаболомикой и данными ATAC-seq для построения многоуровневых моделей.
Perturb-seq улучшает функциональный анализ iPSC-моделей заболеваний, выявляя регуляторные «бутылочные горлышки» в транскриптомном ответе клетки. Это особенно ценно при работе с индуцированными плюрипотентными стволовыми клетками (iPSC), где динамика дифференцировки определяет достоверность модели.
Профессиональный совет: При проектировании Perturb-seq эксперимента включайте негативные контроли с нецелевыми направляющими РНК в каждую партию клеток. Это позволяет корректно разделить технический шум и биологический сигнал при последующем вычислительном анализе.

Интеграция мультиомиксных данных через биомедицинские фундаментальные модели (Foundation Models, FM) значительно усиливает предсказательную силу транскриптомного анализа. Транскриптом описывает текущее состояние клетки, тогда как мультиомиксный контекст объясняет, почему это состояние возникло.
Как транскриптомика помогает преодолевать диагностические трудности при редких заболеваниях?
Диагностика редких генетических болезней остаётся одной из самых сложных задач клинической генетики. Стандартное экзомное секвенирование (WES) выявляет варианты, но не всегда объясняет их функциональные последствия. Варианты неопределённого клинического значения (VUS) составляют значительную долю находок при секвенировании, и именно здесь транскриптомика даёт решающее преимущество.
RNA-seq выявляет функциональные эффекты VUS и сплайсинговых вариантов, невидимых при анализе ДНК. Это означает, что мутация, классифицированная как VUS по геномным данным, может быть переклассифицирована в патогенную или доброкачественную после анализа её влияния на транскрипт.
| Диагностическая задача | Метод | Результат |
|---|---|---|
| Интерпретация VUS | RNA-seq + WGS | Выявление аберрантного сплайсинга или изменения уровня экспрессии |
| Компаунд-гетерозиготность | Фазирование транскриптов | Подтверждение наличия двух патогенных аллелей в транс-конфигурации |
| Нарушения регуляции | ATAC-seq + RNA-seq | Обнаружение изменений в доступности хроматина, влияющих на экспрессию |
| Редкие клеточные фенотипы | scRNA-seq | Идентификация патологических субпопуляций клеток |
Компаунд-гетерозиготность представляет особую сложность: два патогенных варианта в одном гене, расположенных на разных хромосомах, могут быть пропущены при стандартном анализе. Транскриптомный анализ позволяет подтвердить их совместное влияние на функцию гена через изменение уровня или структуры транскрипта.
Изучение редких болезней через геномику показывает, что интеграция RNA-seq с полногеномным секвенированием (WGS) сокращает долю неинтерпретированных вариантов. Это не просто академический прогресс: для пациента это разница между годами диагностических скитаний и конкретным диагнозом.
Профессиональный совет: Для максимальной диагностической отдачи от RNA-seq используйте ткань, наиболее релевантную для предполагаемого заболевания. Транскриптом лимфоцитов крови не отражает специфические нарушения в нейронах или кардиомиоцитах, поэтому iPSC-производные клетки нужного типа дают значительно более информативный результат.
Какие вычислительные методы применяются для анализа транскриптомных данных при редких болезнях?
Вычислительные модели стали неотъемлемой частью транскриптомного анализа в исследовании редких заболеваний. Они позволяют извлекать клинически значимые паттерны из данных, которые человек не способен проанализировать вручную.
Модель «виртуального биохимического двойника» (Virtual Biochemical Twin) строит математическое описание клеточной системы на основе транскриптомных данных. Средний коэффициент корреляции Пирсона r = 0,89 при предсказании реакции органоидов на генетические возмущения подтверждает высокую точность этого подхода. Это означает, что виртуальная модель воспроизводит биологическую реальность с достаточной точностью для предварительного скрининга терапевтических гипотез.
Биомедицинские фундаментальные модели (FM) работают иначе: они предобучаются на миллионах транскриптомных профилей, а затем дообучаются на малых выборках конкретного заболевания. Исследование на 51 пациенте демонстрирует, что интеграция мультиомиксных данных через FM повышает точность прогноза терапевтического ответа даже при ограниченном числе наблюдений. Для редких болезней, где когорты пациентов по определению малы, это принципиально важно.
Ключевые вычислительные подходы в транскриптомном анализе редких заболеваний:
- Дифференциальный анализ экспрессии (DESeq2, edgeR): выявляет гены с аномальной активностью по сравнению с контролем.
- Анализ траекторий (Monocle, Velocity): реконструирует динамику клеточной дифференцировки и выявляет точки нарушения при патологии.
- Сетевой анализ (WGCNA): строит модули совместно регулируемых генов и идентифицирует ключевые регуляторные узлы.
- Фундаментальные модели (Geneformer, scGPT): применяют трансформерную архитектуру к транскриптомным данным для предсказания клеточных состояний.
- AlphaGenome: анализирует длинные участки ДНК и предсказывает влияние мутаций на экспрессию генов.
AlphaGenome сужает круг поиска мутаций, нарушающих сплайсинг или регуляторные участки, что особенно ценно при анализе некодирующих вариантов в редких заболеваниях. Однако ИИ-инструменты ускоряют первичный анализ, но клиническая реализация требует экспериментального подтверждения.
Переход к предиктивной биологии на основе интеграции транскриптомных данных с динамическими моделями открывает путь к персонализированной медицине редких заболеваний, где терапевтические решения принимаются на основе функционального портрета конкретного пациента, а не усреднённых популяционных данных.
Роль молекулярной биологии в редких болезнях трансформируется именно через эти вычислительные методы: от описания к предсказанию, от диагностики к прогнозированию терапевтического ответа.
Какие ограничения существуют при применении транскриптомики в моделировании редких болезней?
Транскриптомика не лишена ограничений, и понимание этих ограничений определяет качество исследования. Игнорирование их приводит к ложным выводам и неверным терапевтическим гипотезам.
-
Недостаток данных по редким клеточным состояниям. Наборы данных для редких заболеваний часто ограничены, а многие предиктивные модели требуют подтверждения лабораторными тестами. Алгоритмы, обученные на распространённых заболеваниях, плохо переносятся на редкие клеточные фенотипы.
-
Тканевая специфичность транскриптома. Транскриптом клетки зависит от её типа, стадии дифференцировки и микросреды. Анализ доступной ткани (например, крови) не всегда отражает патологические изменения в поражённом органе.
-
Техническая вариабельность. Протоколы выделения РНК, глубина секвенирования и методы нормализации данных влияют на воспроизводимость результатов между лабораториями. Это особенно критично при сравнении данных из разных центров.
-
Необходимость лабораторной верификации ИИ-предсказаний. Результаты вычислительных моделей необходимо верифицировать ПЦР в реальном времени и функциональными тестами из-за возможного несоответствия данных и редких клеточных фенотипов. Вычислительная модель генерирует гипотезу, а не диагноз.
-
Динамичность транскриптома. Экспрессия генов меняется в ответ на стресс, лечение и стадию болезни. Однократный срез транскриптома может не отражать хроническое патологическое состояние.
Перспективы преодоления этих ограничений связаны с несколькими направлениями. Динамическое моделирование с использованием Perturb-seq позволяет отслеживать транскриптомные изменения во времени, а не фиксировать единственный момент. Мультиомиксная интеграция компенсирует слабые стороны каждого отдельного метода. Международные консорциумы, такие как Undiagnosed Diseases Network (UDN) и EURO-RDI, накапливают транскриптомные данные по редким заболеваниям, постепенно решая проблему малых выборок.
Понимание сложностей диагностики редких болезней помогает правильно выстраивать исследовательский протокол: транскриптомика усиливает геномный анализ, но не заменяет его.
Ключевые выводы
Транскриптомика обеспечивает функциональную интерпретацию генетических вариантов и строит точные клеточные модели редких заболеваний там, где ДНК-анализ останавливается.
| Пункт | Подробности |
|---|---|
| Диагностический разрыв | До 40% пациентов с моногенными болезнями не получают диагноза без транскриптомного анализа. |
| Ключевые технологии | scRNA-seq, пространственная транскриптомика и Perturb-seq раскрывают клеточную архитектуру болезни. |
| Вычислительные модели | Виртуальные биохимические двойники с r = 0,89 позволяют предсказывать терапевтический ответ на малых выборках. |
| Ограничения метода | Редкие клеточные фенотипы требуют лабораторной верификации всех ИИ-предсказаний. |
| Интеграция данных | Объединение RNA-seq с WGS и мультиомиксными данными повышает точность диагностики и прогноза. |
Транскриптомика и редкие болезни: взгляд из лаборатории
За годы работы с транскриптомными данными в контексте редких генетических заболеваний я пришёл к одному неочевидному выводу: самая большая проблема в этой области не техническая, а интерпретационная.
Технологии scRNA-seq и пространственной транскриптомики достигли зрелости. Стоимость секвенирования продолжает падать. Вычислительные инструменты становятся доступнее. Но разрыв между «мы видим аномальный транскриптомный паттерн» и «мы понимаем, что с этим делать клинически», остаётся огромным.
Я наблюдал, как исследовательские группы тратили месяцы на генерацию красивых UMAP-визуализаций, не имея чёткого плана функциональной валидации. Транскриптомные данные соблазняют своей детальностью. Но детальность без биологической гипотезы превращается в дорогостоящий каталог наблюдений.
Практический вывод, который я вынес из работы с iPSC-моделями: начинайте с клинического вопроса, а не с технологии. Какой именно механизм вы хотите понять? Какое терапевтическое решение должен поддержать анализ? Только с этой отправной точки транскриптомика раскрывает свой потенциал в полной мере. Роль молекулярной биологии в редких болезнях реализуется именно тогда, когда молекулярные данные отвечают на конкретный клинический вопрос, а не существуют сами по себе.
Междисциплинарное сотрудничество между биоинформатиками, клиническими генетиками и специалистами по клеточной биологии не просто желательно. Без него транскриптомный анализ редких заболеваний теряет половину своей ценности.
— John
Hopeatrarelabs: моделирование редких заболеваний на основе транскриптомики
Hopeatrarelabs специализируется на создании персонализированных моделей ультраредких и недиагностированных генетических заболеваний на основе клеток пациента. Платформа объединяет iPSC-технологии, CRISPR-редактирование и транскриптомный анализ для построения функциональных клеточных моделей, которые затем используются для параллельного скрининга тысяч одобренных FDA препаратов, антисмысловых олигонуклеотидов (ASO) и вариантов генной терапии.
Для исследователей и клиницистов, работающих с редкими генетическими болезнями, Hopeatrarelabs предлагает доступ к базе знаний по редким болезням, которая поддерживает разработку исследовательских программ. Платформа персонализированного моделирования ориентирована на случаи, где стандартные диагностические и терапевтические подходы исчерпали свои возможности.
Часто задаваемые вопросы
Что такое транскриптомика в контексте редких болезней?
Транскриптомика анализирует всю совокупность РНК-молекул клетки для изучения активности генов. При редких заболеваниях она позволяет интерпретировать функциональные последствия генетических мутаций, невидимые при анализе ДНК.
Почему стандартного секвенирования недостаточно для диагностики?
До 40% пациентов с подозрением на моногенные заболевания не получают окончательного диагноза после стандартного секвенирования. Транскриптомный анализ выявляет аберрантный сплайсинг и изменения экспрессии, которые объясняют патогенность вариантов неопределённого значения.
Как Perturb-seq используется в моделировании редких заболеваний?
Perturb-seq объединяет CRISPR-скрининг с одноклеточным транскриптомным профилированием: каждая клетка получает конкретное генетическое возмущение, а её транскриптомный ответ фиксируется одновременно. Это позволяет выявлять регуляторные нарушения в iPSC-моделях заболеваний.
Что такое виртуальный биохимический двойник?
Виртуальный биохимический двойник строит математическую модель клеточной системы на основе транскриптомных данных. Коэффициент корреляции r = 0,89 при предсказании реакции органоидов подтверждает достаточную точность для предварительного скрининга терапевтических гипотез.
Нужна ли лабораторная верификация результатов ИИ-моделей?
Да. Результаты вычислительных моделей, включая предсказания AlphaGenome и фундаментальных моделей, требуют верификации ПЦР в реальном времени и функциональными тестами. Вычислительная модель генерирует гипотезу, а не клинический диагноз.