
CRISPR aplicado al modelado de enfermedades monogénicas es la estrategia más directa para reproducir mutaciones causantes de patología en sistemas celulares y animales controlados. La edición génica dirigida, término estándar en la literatura biomédica, permite introducir o corregir variantes patogénicas con una resolución que ninguna técnica anterior igualó. Esto convierte a CRISPR en una plataforma dual: herramienta de investigación patogénica y banco de pruebas terapéutico. Las enfermedades monogénicas, causadas por mutaciones en un único gen, son el escenario ideal para esta tecnología, ya que el objetivo genético está bien definido. El Dr. Ignacio Pérez de Castro del ISCIII describe CRISPR como un «avatar» terapéutico que permite predecir eficacia y toxicidad antes de cualquier ensayo en humanos.
¿Qué herramientas CRISPR son más adecuadas para aplicar en un modelo de enfermedad monogénica?
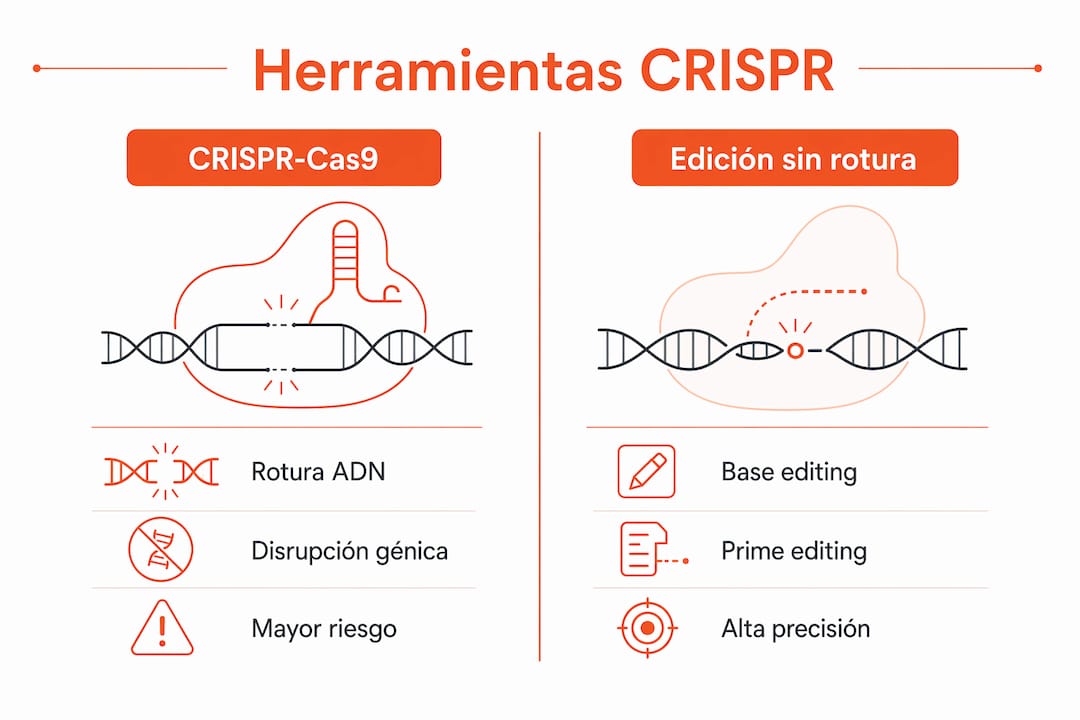
La elección de la herramienta CRISPR determina la fidelidad del modelo y la seguridad del abordaje terapéutico. No todas las variantes de edición génica son equivalentes, y la mutación diana dicta cuál es la más apropiada.
CRISPR-Cas9 es la plataforma más consolidada. Genera roturas de doble hebra en el ADN y permite tanto la disrupción génica como la inserción de secuencias mediante recombinación homóloga. Su eficiencia es alta, pero las roturas de doble hebra introducen riesgo de reordenamientos cromosómicos y efectos fuera de diana, especialmente en modelos in vivo.

La edición de bases (base editing) modifica nucleótidos individuales sin generar roturas. Es la opción preferida para mutaciones puntuales, como las que causan la enfermedad renal poliquística autosómica dominante (ERPAD). Un estudio preclínico en Mayo Clinic demostró que la corrección del gen PKD1 mediante edición de bases ralentizó el crecimiento de quistes renales y mejoró la función renal y hepática tras una sola dosis. Ese resultado con una intervención única tiene implicaciones directas para el diseño de ensayos clínicos.
Prime editing amplía el rango de correcciones posibles: inserciones, deleciones y sustituciones de cualquier tipo sin rotura de doble hebra. PERT (Programmable endogenous riboregulator technology) actúa a nivel de ARN, convirtiendo un ARNt endógeno en un supresor optimizado para leer codones de terminación prematura. PERT restaura la traducción proteica de forma permanente sin toxicidad ni alteraciones transcriptómicas significativas, lo que lo convierte en una opción especialmente atractiva para mutaciones sin sentido.
| Técnica | Tipo de mutación diana | Rotura de ADN | Perfil de seguridad |
|---|---|---|---|
| CRISPR-Cas9 | Disrupción, inserción, deleción | Sí (doble hebra) | Moderado |
| Edición de bases | Mutaciones puntuales | No | Alto |
| Prime editing | Inserciones, deleciones, sustituciones | No | Alto |
| PERT | Mutaciones sin sentido (codón stop prematuro) | No | Muy alto |
Consejo profesional: Seleccione prime editing o edición de bases cuando la mutación diana sea una variante puntual conocida. Reserve CRISPR-Cas9 para disrupciones génicas completas o modelos knock-in complejos donde la rotura de doble hebra sea aceptable.
¿Cómo implementar modelos celulares y animales con CRISPR para enfermedades monogénicas?
El diseño experimental riguroso es la diferencia entre un modelo que recapitula la enfermedad y uno que introduce artefactos. El proceso sigue una secuencia lógica que conviene respetar.
Diseño y validación de la guía CRISPR
- Selección del ARN guía (sgRNA). Diseñe el sgRNA con herramientas bioinformáticas validadas como CRISPOR o Benchling. Priorice secuencias con alta especificidad y baja puntuación de efectos fuera de diana. Valide in silico antes de cualquier experimento celular.
- Validación en células primarias o líneas celulares. Transfecte el complejo ribonucleoproteico (RNP) en células HEK293T o en células primarias del tejido relevante. Cuantifique la eficiencia de edición mediante secuenciación de amplicones (amplicon-seq) o TIDE.
- Confirmación de la edición en el alelo correcto. Use secuenciación de Sanger o secuenciación de nueva generación (NGS) para verificar que la mutación introducida replica exactamente la variante patogénica. Descarte mosaicismos no deseados.
- Generación del modelo animal. Inyecte el complejo CRISPR en cigotos murinos o use vectores virales para edición postnatal. Los vectores adenoasociados (AAV) son el estándar para administración in vivo por su tropismo tisular selectivo y bajo perfil inmunogénico.
- Caracterización fenotípica. Evalúe el fenotipo a nivel molecular (Western blot, RT-qPCR, proteómica), histológico e funcional. Compare con controles no editados del mismo fondo genético.
Consideraciones para modelos iPSC
Los modelos basados en células madre pluripotentes inducidas (iPSC) ofrecen una ventaja única: derivan de células del propio paciente. Esto permite replicar el contexto genómico completo, incluidas variantes modificadoras. La diferenciación de iPSC editadas hacia el tipo celular relevante (cardiomiocitos, neuronas, células renales) genera modelos con alta relevancia traslacional. La edición ex vivo sigue siendo el estándar de seguridad, aunque los avances en nanopartículas lipídicas y vectores AAV optimizados amplían las opciones para edición in vivo en órganos sólidos.
- Confirme pluripotencia tras la edición mediante marcadores OCT4, SOX2 y NANOG.
- Verifique integridad cromosómica con cariotipo o secuenciación de genoma completo.
- Incluya controles isogénicos: líneas iPSC del mismo donante sin la mutación introducida.
¿Cuáles son los avances recientes en modelado CRISPR de enfermedades monogénicas?
Los resultados preclínicos de 2025 y 2026 redefinen los umbrales de eficacia que se consideraban necesarios para el éxito terapéutico.
El caso más ilustrativo es el de la distrofia muscular congénita por mutación en LMNA (L-CMD). Investigadores del ISCIII aplicaron CRISPR-Cas9 con vectores AAV en un modelo murino y obtuvieron un incremento de supervivencia superior al 20 % con menos del 10 % de cardiomiocitos editados. Ese dato invierte una asunción central en el campo: no se necesita corregir la mayoría de las células para obtener beneficio clínico significativo.
«No se requiere corrección completa para eficacia terapéutica. Mejoras significativas se logran con edición de menos del 10 % de las células diana, lo que reduce la presión técnica sobre los modelos animales y acelera la validación experimental.»
ISCIII, estudio preclínico en L-CMD, 2026
El estudio de Mayo Clinic en ERPAD refuerza esta tendencia. La edición de bases sobre el gen PKD1 en una sola dosis detuvo la progresión quística y mejoró parámetros de función renal y hepática en modelos preclínicos. La relevancia de este resultado radica en que PKD1 es uno de los genes más grandes del genoma humano, lo que históricamente complicaba su abordaje con vectores virales convencionales.
En el plano regulatorio, la FDA estableció un marco específico para enfermedades ultra-raras que permite ensayos clínicos desde 5–10 participantes para plataformas de edición genómica personalizadas. Además, el concepto de «mecanismo plausible» permite agrupar múltiples editores genéticos dirigidos a mutaciones de un solo gen bajo una única solicitud de aprobación clínica. Para los investigadores que trabajan en criterios de designación huérfana, este marco reduce sustancialmente la barrera regulatoria hacia el ensayo clínico.
¿Qué desafíos deben considerarse al aplicar CRISPR en modelos de enfermedades monogénicas?
La edición génica en modelos preclínicos presenta obstáculos técnicos que, si no se gestionan desde el diseño experimental, comprometen la reproducibilidad y la traslación clínica.
- Eficacia de edición variable. La eficiencia del sgRNA depende de la accesibilidad de la cromatina en el locus diana. Loci en heterocromatina densa pueden mostrar eficiencias inferiores al 5 %, insuficientes para generar un fenotipo reproducible. Valide siempre la eficiencia en el tipo celular específico del modelo, no solo en líneas de referencia.
- Efectos fuera de diana. CRISPR-Cas9 puede editar secuencias con homología parcial al sgRNA. Use secuenciación de genoma completo o métodos como GUIDE-seq para mapear sitios fuera de diana antes de avanzar a modelos animales.
- Toxicidad del sistema de entrega. Los vectores AAV pueden generar respuesta inmune en modelos murinos inmunocompetentes. Evalúe títulos virales y considere modelos inmunodeficientes para separar toxicidad inmune de toxicidad génica.
- Mosaicismo en modelos animales. La edición en cigotos genera frecuentemente animales mosaico, donde solo una fracción de células porta la mutación. Genotipe múltiples tejidos y seleccione fundadores con edición germinal confirmada.
Las técnicas sin rotura de doble hebra como prime editing y PERT eliminan los riesgos asociados a reordenamientos cromosómicos y reducen la señal de daño al ADN, lo que mejora la viabilidad celular en modelos sensibles. Para enfermedades donde la toxicidad del proceso de edición podría confundir el fenotipo de la enfermedad, esta ventaja es determinante.
Consejo profesional: Incluya siempre un grupo control con el sistema de entrega sin el componente de edición activo. Esto permite distinguir efectos del vector o del proceso de transfección del fenotipo genuinamente atribuible a la mutación.
Puntos clave
Aplicar CRISPR en modelos de enfermedades monogénicas requiere seleccionar la técnica según el tipo de mutación, validar la edición en el contexto celular correcto y asumir que menos del 10 % de edición puede ser suficiente para obtener eficacia terapéutica significativa.
| Punto | Detalles |
|---|---|
| Elección de técnica | Use edición de bases o prime editing para mutaciones puntuales; reserve Cas9 para disrupciones complejas. |
| Validación de edición | Confirme la variante con NGS en el tipo celular diana antes de generar modelos animales. |
| Umbral de eficacia | Menos del 10 % de células editadas puede producir beneficio terapéutico significativo en modelos murinos. |
| Marco regulatorio | La FDA permite ensayos desde 5–10 participantes para plataformas de edición génica en enfermedades ultra-raras. |
| Control experimental | Incluya controles isogénicos y grupos con vector sin edición activa para aislar el fenotipo de la enfermedad. |
CRISPR como plataforma de medicina de precisión: una perspectiva desde la práctica
Lo que más me ha sorprendido trabajando con modelos CRISPR en enfermedades monogénicas no es la precisión de la edición. Es el cambio de mentalidad que exige. Durante años, el estándar implícito fue «corregir el máximo posible». Los datos del ISCIII en L-CMD demuestran que ese objetivo era innecesariamente ambicioso, y en algunos casos, contraproducente.
El verdadero valor de CRISPR en este campo no es solo terapéutico. Es epistémico. Cuando introduces una mutación específica en un modelo isogénico y observas el fenotipo resultante, estás haciendo algo que la genética clásica no podía: establecer causalidad, no correlación. Eso cambia cómo se interpreta la patogénesis y cómo se diseñan los ensayos de fármacos.
Lo que veo como el próximo salto real no es una nueva nucleasa. Es la integración de modelos CRISPR con plataformas de cribado de alto rendimiento, como las que usa Hopeatrarelabs para opciones terapéuticas en enfermedades genéticas sin cura. Un modelo bien validado es inútil si no se conecta con un sistema que pueda extraer información terapéutica de él a escala. La combinación de iPSC editadas con CRISPR y cribado paralelo de miles de compuestos es donde la investigación en enfermedades monogénicas tiene más probabilidades de producir resultados clínicamente relevantes en los próximos cinco años.
— John
Hopeatrarelabs y el modelado CRISPR en enfermedades monogénicas
Hopeatrarelabs trabaja específicamente en el espacio donde la edición génica y el modelado de enfermedades ultra-raras convergen. Su plataforma genera modelos derivados de células del propio paciente mediante iPSC y CRISPR, y los conecta directamente con cribados paralelos de fármacos aprobados por la FDA, oligonucleótidos antisentido personalizados y evaluación de terapias génicas.
Para investigadores que necesitan pasar del modelo validado a la identificación de opciones terapéuticas con rigor científico, el recurso especializado de Hopeatrarelabs ofrece acceso a programas estructurados para enfermedades raras y ultra-raras. La plataforma principal de medicina de precisión e iPSC está diseñada para acortar el tiempo entre la caracterización genética y la identificación de una opción terapéutica viable.
Preguntas frecuentes
¿Qué es aplicar CRISPR en un modelo de enfermedad monogénica?
Consiste en introducir o corregir una mutación causante de enfermedad en células o animales mediante edición génica dirigida, para replicar la patología y evaluar opciones terapéuticas en un sistema controlado.
¿Qué porcentaje de edición celular se necesita para eficacia terapéutica?
Estudios preclínicos del ISCIII en distrofia muscular L-CMD demostraron mejoras de supervivencia superiores al 20 % con menos del 10 % de cardiomiocitos editados. La corrección total no es necesaria.
¿Cuál es la diferencia entre edición ex vivo e in vivo en modelos CRISPR?
La edición ex vivo modifica células fuera del organismo antes de reintroducirlas, con mayor control y seguridad. La edición in vivo administra el sistema CRISPR directamente al organismo mediante vectores como AAV o nanopartículas lipídicas.
¿Qué ventaja ofrece prime editing frente a CRISPR-Cas9 en enfermedades monogénicas?
Prime editing no genera roturas de doble hebra, lo que reduce el riesgo de reordenamientos cromosómicos y efectos fuera de diana. Permite además corregir inserciones, deleciones y sustituciones de cualquier tipo con mayor precisión.
¿Cómo afecta el marco regulatorio de la FDA al desarrollo de modelos CRISPR terapéuticos?
La FDA permite ensayos clínicos desde 5–10 participantes para plataformas de edición genómica en enfermedades ultra-raras, y agrupa editores genéticos bajo el concepto de «mecanismo plausible», reduciendo los requisitos para avanzar desde el modelo preclínico al ensayo clínico.