
Un medicamento huérfano es un fármaco destinado a tratar enfermedades raras que afectan a un número muy reducido de personas y para las que no existen alternativas terapéuticas satisfactorias. Los criterios que definen qué son criterios orphan drug enfermedades raras están establecidos por ley en la Unión Europea y en Estados Unidos, con umbrales de prevalencia precisos y requisitos clínicos específicos. Entender estos criterios no es solo útil para investigadores: para una familia que busca opciones de tratamiento, saber cómo funciona este sistema puede marcar la diferencia entre encontrar una vía o perderse en el laberinto regulatorio.
¿Cuáles son los criterios reguladores para la designación de medicamentos huérfanos?
La definición de medicamentos huérfanos varía según la jurisdicción, pero comparte una lógica común: compensar la falta de incentivos comerciales para desarrollar fármacos destinados a poblaciones muy pequeñas.
En la Unión Europea, el Reglamento (CE) 141/2000 establece que una enfermedad debe afectar a no más de 5 personas por cada 10.000 para que un medicamento pueda recibir la designación huérfana. Además, el solicitante debe demostrar que no existe ninguna alternativa satisfactoria autorizada, o bien que el nuevo fármaco ofrece un beneficio significativo frente a las opciones existentes. La evaluación la realiza el Comité de Medicamentos Huérfanos de la Agencia Europea de Medicamentos (EMA/COMP).

En Estados Unidos, la Orphan Drug Act define la elegibilidad de forma diferente: una enfermedad califica si afecta a menos de 200.000 personas en el país. Este umbral numérico absoluto contrasta con el criterio europeo de prevalencia relativa. La FDA gestiona el proceso y otorga la designación con incentivos propios.
La tabla siguiente resume las diferencias principales entre ambos marcos:
| Criterio | Unión Europea | Estados Unidos |
|---|---|---|
| Umbral de prevalencia | ≤5 por cada 10.000 habitantes | Menos de 200.000 personas en total |
| Organismo evaluador | EMA / COMP | FDA |
| Requisito clínico adicional | Beneficio significativo o ausencia de alternativa | Ausencia de alternativa o beneficio clínico plausible |
| Exclusividad de mercado | 10 años (12 en pediatría) | 7 años |
| Marco legal | Reglamento (CE) 141/2000 | Orphan Drug Act (1983) |
Un matiz que muchos pasan por alto: la indicación específica del medicamento determina qué población se considera para calcular la prevalencia. Un mismo compuesto puede ser huérfano para un subtipo de una enfermedad y no serlo para otro. Esto tiene consecuencias directas en la estrategia de desarrollo clínico.
Consejo profesional: Si usted está preparando una solicitud de designación, defina la indicación con la mayor precisión posible desde el inicio. Una indicación demasiado amplia puede exceder el umbral de prevalencia y perder la elegibilidad huérfana.
¿Qué beneficios regulatorios reciben los medicamentos con designación huérfana?
Los incentivos para el desarrollo de fármacos huérfanos existen porque el mercado por sí solo no justifica la inversión. Sin ellos, la mayoría de los proyectos para enfermedades poco comunes nunca llegarían a ensayo clínico.
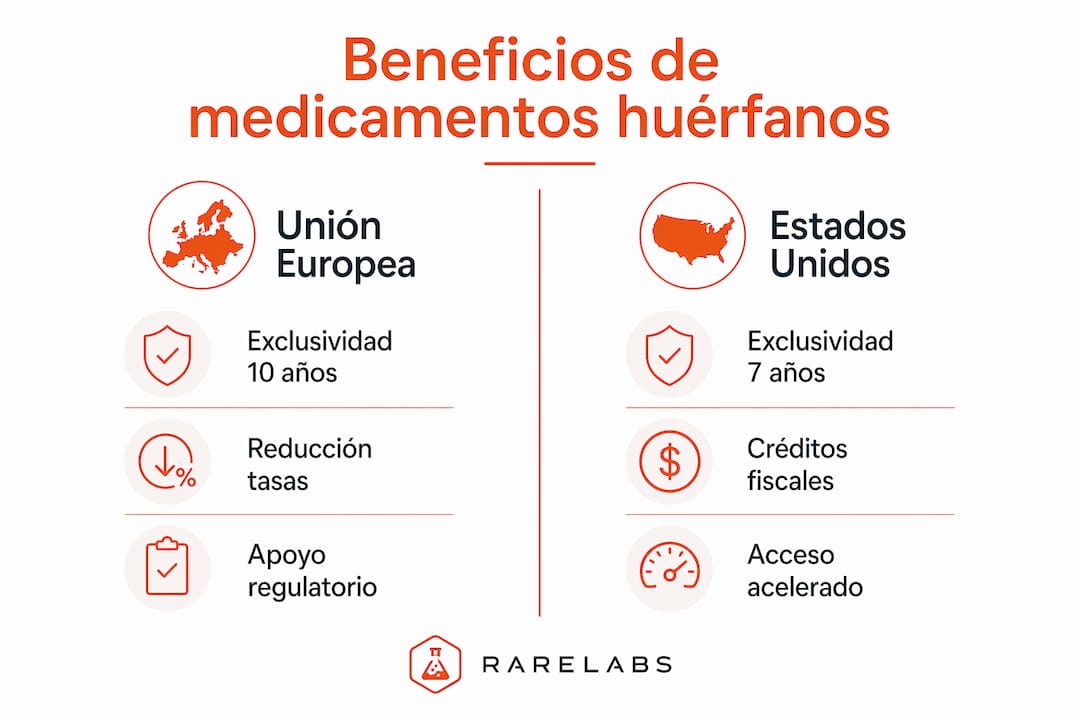
Los principales beneficios que otorga la designación huérfana en Europa y Estados Unidos son:
- Exclusividad de mercado. En la UE, la exclusividad comercial es de 10 años (12 años si el medicamento está indicado en pediatría). En EE.UU., la Orphan Drug Act garantiza 7 años. Durante ese período, la autoridad reguladora no puede aprobar un medicamento similar para la misma indicación.
- Reducción o exención de tasas regulatorias. La EMA ofrece exenciones significativas en las tasas de evaluación para solicitantes con designación huérfana, lo que reduce el coste del proceso de autorización.
- Asesoramiento científico prioritario. Tanto la EMA como la FDA ofrecen asesoramiento científico y orientación regulatoria específica para proyectos huérfanos, lo que acelera el diseño de los ensayos clínicos.
- Programas acelerados de evaluación. La FDA dispone del programa Fast Track y la EMA del procedimiento PRIME (Priority Medicines), ambos accesibles para medicamentos huérfanos con alto potencial terapéutico.
- Subvenciones y apoyo financiero. En Europa, programas como Horizon Europe financian parte de la investigación en enfermedades raras. En EE.UU., la FDA ofrece créditos fiscales por gastos en ensayos clínicos huérfanos.
Estos incentivos son esenciales para compensar el alto riesgo financiero que supone generar evidencia clínica en poblaciones tan pequeñas. Sin ellos, el coste por paciente en un ensayo clínico para una enfermedad ultra-rara haría inviable cualquier proyecto privado.
Consejo profesional: La designación huérfana no garantiza la aprobación del medicamento ni su financiación pública. Planifique desde el primer día la estrategia de acceso al mercado como un proceso paralelo al desarrollo clínico, no como una etapa posterior.
¿Cómo se aplican en la práctica los criterios y qué retos enfrentan investigadores y familias?
Obtener la designación huérfana es solo el primer paso. La aplicación real de los criterios para enfermedades raras implica una carga documental y estratégica que muchos subestiman.
El proceso exige un dossier técnico riguroso que incluye:
- Evidencia epidemiológica. El solicitante debe aportar datos que justifiquen la rareza de la enfermedad en la población de referencia. Esto requiere revisar registros epidemiológicos, publicaciones científicas y bases de datos de enfermedades raras como Orphanet.
- Justificación clínica del beneficio. Si ya existe algún tratamiento autorizado, el dossier debe demostrar que el nuevo fármaco ofrece un beneficio significativo. Esta justificación es, según la EMA, el principal desafío para los solicitantes.
- Definición precisa de la indicación. Como se explicó antes, la indicación determina la población elegible. Un error en este punto puede invalidar toda la solicitud.
- Análisis de alternativas terapéuticas. El dossier debe incluir una revisión exhaustiva de los tratamientos existentes y argumentar por qué son insatisfactorios para la población objetivo.
Para las familias de pacientes, el reto más frecuente es confundir la designación huérfana con el acceso inmediato al medicamento. El estatus huérfano no garantiza acceso inmediato: la autorización de comercialización y la financiación pública son procesos independientes y posteriores. En España, las demoras en financiación pública de medicamentos huérfanos pueden alcanzar los 23–24 meses desde la autorización europea. Eso significa que un medicamento aprobado por la EMA puede tardar casi dos años en estar disponible para un paciente en España.
Para los investigadores, la dificultad adicional está en los fondos para investigación pediátrica rara y en el diseño de ensayos clínicos con muestras muy reducidas, donde los métodos estadísticos convencionales no son aplicables.
¿Cuál es la diferencia entre designación huérfana y autorización de comercialización?
La designación huérfana y la autorización de comercialización son dos procesos completamente distintos. Confundirlos es el error más común entre familias e investigadores que se acercan por primera vez a este sistema.
La designación huérfana es un estatus regulatorio que habilita al desarrollador para acceder a los incentivos descritos anteriormente. No implica que el medicamento sea seguro, eficaz ni que esté disponible para los pacientes. Es, en esencia, una etiqueta que dice: "este proyecto merece apoyo especial porque aborda una necesidad médica no cubierta."
La autorización de comercialización, en cambio, certifica que el medicamento ha superado los ensayos clínicos y cumple los estándares de seguridad y eficacia exigidos. En Europa, la EMA otorga la Autorización de Comercialización Centralizada. En EE.UU., la FDA emite la New Drug Application (NDA) o la Biologics License Application (BLA).
La tabla siguiente ilustra el recorrido completo de un medicamento huérfano:
| Etapa | Descripción | Organismo responsable |
|---|---|---|
| Designación huérfana | Reconocimiento del potencial terapéutico en enfermedad rara | EMA (Europa) / FDA (EE.UU.) |
| Ensayos clínicos | Demostración de seguridad y eficacia en la indicación | Promotor del estudio |
| Autorización de comercialización | Aprobación para venta y uso clínico | EMA / FDA |
| Financiación pública | Decisión de cobertura por sistemas nacionales de salud | Ministerios / agencias nacionales |
| Acceso al paciente | Dispensación efectiva en centros sanitarios | Sistema sanitario nacional |
El papel de la EMA y la FDA es central en las primeras etapas, pero el acceso real al paciente depende de decisiones nacionales. En España, el Ministerio de Sanidad y el Consejo Interterritorial del Sistema Nacional de Salud son los actores clave en la etapa de financiación. Para una familia, entender este mapa de responsabilidades es tan importante como conocer los criterios científicos del medicamento.
Puntos clave
Los criterios para la designación de medicamentos huérfanos definen una vía regulatoria específica que no equivale a acceso inmediato al tratamiento, sino al inicio de un proceso con etapas diferenciadas.
| Punto | Detalles |
|---|---|
| Umbral de prevalencia en la UE | Una enfermedad debe afectar a no más de 5 personas por cada 10.000 para calificar. |
| Umbral en EE.UU. | La Orphan Drug Act exige que la enfermedad afecte a menos de 200.000 personas en total. |
| Designación no es autorización | La designación huérfana habilita incentivos, pero no garantiza acceso ni aprobación del fármaco. |
| Exclusividad de mercado | La UE otorga 10 años de exclusividad comercial; EE.UU. otorga 7 años. |
| Acceso en España | Las demoras en financiación pública pueden superar los 23 meses tras la autorización europea. |
Lo que nadie explica sobre los medicamentos huérfanos
Llevo años trabajando en el entorno de las enfermedades raras y hay algo que me sigue sorprendiendo: la mayoría de los artículos sobre medicamentos huérfanos explican los criterios regulatorios con precisión, pero casi ninguno prepara a las familias para lo que viene después de la designación.
El sistema de incentivos que creó la Orphan Drug Act en 1983 y que Europa adoptó con el Reglamento 141/2000 fue un avance real. Antes de esas leyes, desarrollar un fármaco para 5.000 pacientes era económicamente inviable para cualquier empresa. Los incentivos cambiaron esa ecuación. Hoy existen más de 200 medicamentos huérfanos autorizados en Europa. Eso no habría ocurrido sin ese marco regulatorio.
Pero el sistema tiene una grieta que afecta especialmente a los pacientes en España y en otros mercados con procesos de financiación lentos. Una familia puede ver cómo la EMA aprueba el medicamento que necesita su hijo y luego esperar casi dos años para que ese fármaco llegue a la farmacia hospitalaria. Ese intervalo no es un fallo técnico: es una consecuencia estructural de tener regulación centralizada europea y financiación descentralizada nacional.
Lo que recomiendo a investigadores y familias es tratar el proceso de acceso como una carrera paralela al desarrollo clínico, no como una etapa posterior. Documentar la carga de enfermedad desde el inicio, establecer contacto con las agencias de evaluación de tecnologías sanitarias y construir evidencia de valor real para el sistema de salud son pasos que deben comenzar mucho antes de la autorización. El marco regulatorio da las herramientas. Usarlas bien requiere estrategia.
— John
Cómo Hopeatrarelabs apoya la investigación en enfermedades raras
Hopeatrarelabs trabaja directamente con familias, investigadores y socios biofarmacéuticos para acelerar el desarrollo de opciones terapéuticas en enfermedades ultra-raras y sin diagnóstico. Su plataforma combina modelos celulares derivados de células del propio paciente mediante tecnología iPSC y edición génica con CRISPR, con cribados paralelos de miles de fármacos aprobados por la FDA.
Si usted está navegando el proceso de designación huérfana o buscando opciones terapéuticas para una enfermedad sin tratamiento aprobado, el centro de recursos especializados de Hopeatrarelabs ofrece guías, herramientas y orientación científica diseñadas específicamente para este entorno. También puede explorar sus servicios de medicina de precisión para enfermedades ultra-raras y conocer cómo un modelo celular personalizado puede abrir nuevas vías de investigación para su caso concreto.
Preguntas frecuentes
¿Qué es la definición de medicamento huérfano?
Un medicamento huérfano es un fármaco desarrollado para tratar enfermedades raras que afectan a muy pocas personas y para las que no existen alternativas terapéuticas satisfactorias. En la UE, el umbral es de 5 afectados por cada 10.000 habitantes; en EE.UU., menos de 200.000 personas en total.
¿La designación huérfana garantiza el acceso al medicamento?
No. La designación huérfana es un estatus regulatorio que habilita incentivos para el desarrollo, pero no implica autorización de comercialización ni financiación pública. En España, el acceso efectivo puede demorarse más de 23 meses tras la aprobación europea.
¿Cuáles son los principales beneficios de los fármacos huérfanos para los desarrolladores?
Los beneficios incluyen exclusividad de mercado (10 años en la UE, 7 en EE.UU.), exención o reducción de tasas regulatorias, asesoramiento científico prioritario de la EMA o la FDA, y acceso a programas acelerados como PRIME o Fast Track.
¿Cómo funcionan los medicamentos huérfanos en la práctica clínica?
Un medicamento huérfano sigue el mismo proceso de ensayos clínicos que cualquier otro fármaco, pero con adaptaciones metodológicas para poblaciones pequeñas. Tras la autorización, su dispensación se realiza habitualmente en centros hospitalarios especializados en enfermedades raras.
¿Qué diferencia hay entre una enfermedad rara y una enfermedad poco común?
Ambos términos se usan de forma intercambiable, pero "enfermedad rara" es el término regulatorio oficial en Europa. La Unión Europea define las enfermedades raras como aquellas con una prevalencia inferior a 5 casos por cada 10.000 personas, independientemente de su denominación coloquial.