
Eine Monogenerkrankung ist eine gesundheitliche Störung, die durch eine Mutation in genau einem einzigen Gen entsteht und den klassischen Mendelschen Vererbungsregeln folgt. Der Fachbegriff lautet monogene Erkrankung oder monogenetische Erkrankung. Beide Begriffe beschreiben dasselbe Prinzip: Ein defektes Gen unterbricht die Produktion eines wichtigen Proteins oder Enzyms, was zu einem charakteristischen Krankheitsbild führt. Wer verstehen will, was eine Monogenerkrankung ist, muss drei Vererbungsmuster kennen: autosomal-dominant, autosomal-rezessiv und X-chromosomal. Moderne Sequenzierverfahren wie die Exom- und Genomsequenzierung haben die Diagnose dieser Erkrankungen grundlegend verändert und ermöglichen heute Aufklärungsraten, die früher undenkbar waren.
Was ist eine Monogenerkrankung und wie entsteht sie?
Eine monogene Erkrankung ist definiert als Störung, bei der die Ursache vollständig in der Veränderung eines einzelnen Gens liegt. Das betroffene Gen liefert normalerweise die Bauanleitung für ein bestimmtes Protein. Fehlt dieses Protein oder funktioniert es nicht richtig, gerät ein biologischer Prozess aus dem Gleichgewicht.
Die Mutation kann vererbt sein oder neu entstehen, also als sogenannte de-novo-Mutation auftreten. Bei einer de-novo-Mutation tragen die Eltern das veränderte Gen nicht, dennoch erkrankt das Kind. Das ist für viele Familien zunächst schwer zu verstehen, weil keine Familiengeschichte auf eine Erkrankung hingedeutet hat.

Monogene Erkrankungen unterscheiden sich grundlegend von multifaktoriellen Erkrankungen wie Typ-2-Diabetes oder Bluthochdruck. Bei multifaktoriellen Erkrankungen wirken viele Gene zusammen, dazu kommen Umweltfaktoren. Bei monogenen Erkrankungen reicht eine einzige Genveränderung aus, um die Krankheit auszulösen. Die Grenzen zwischen monogenen und multifaktoriellen Erkrankungen sind allerdings fließend, da genetische Modifikatoren oft den Schweregrad beeinflussen.
Wie funktioniert die Vererbung bei monogenen Erkrankungen?
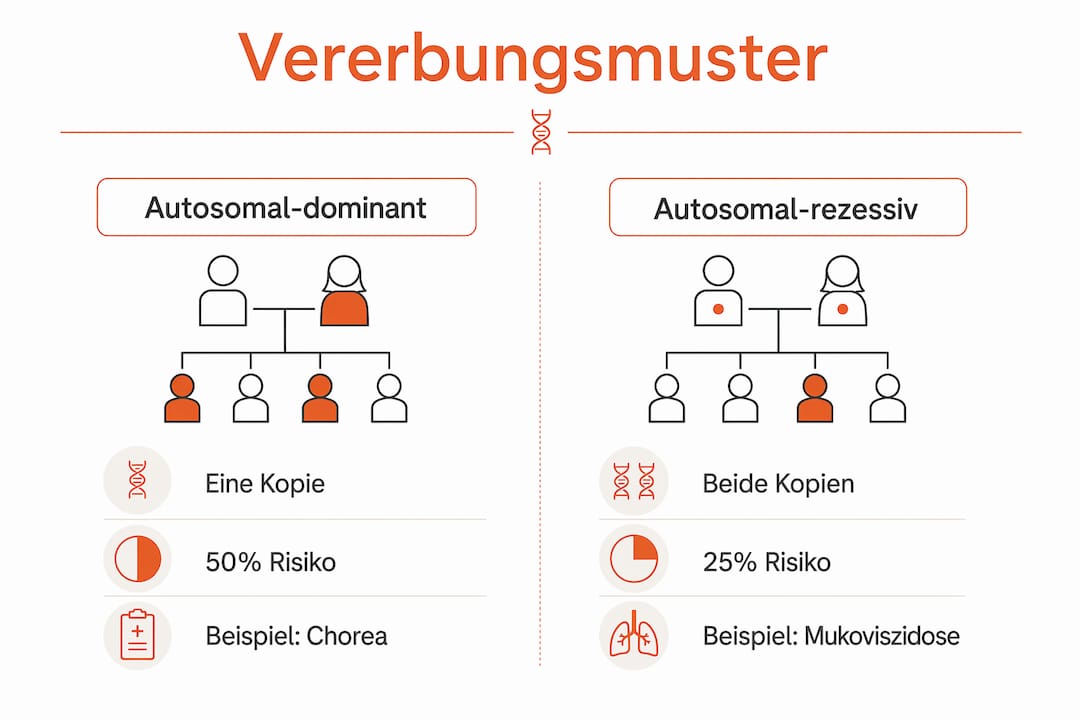
Monogene Erkrankungen folgen den Mendelschen Vererbungsregeln und lassen sich in drei Hauptmuster einteilen. Jedes Muster hat andere Konsequenzen für das Erkrankungsrisiko der Nachkommen.
Autosomal-dominant: Eine einzige veränderte Kopie des Gens reicht aus, um die Erkrankung auszulösen. Das Risiko für Kinder eines betroffenen Elternteils liegt bei 50 % pro Schwangerschaft. Chorea Huntington ist das bekannteste Beispiel: Wer die Mutation trägt, erkrankt im Laufe des Lebens mit hoher Wahrscheinlichkeit.
Autosomal-rezessiv: Beide Kopien des Gens müssen verändert sein, damit die Erkrankung ausbricht. Träger einer einzigen Kopie sind gesund, können die Mutation aber weitergeben. Bei Mukoviszidose beträgt das Erkrankungsrisiko 25 % pro Schwangerschaft, wenn beide Elternteile Anlageträger sind.
X-chromosomal: Das veränderte Gen liegt auf dem X-Chromosom. Männer, die nur ein X-Chromosom haben, erkranken bei einer Mutation häufig direkt. Frauen mit zwei X-Chromosomen sind oft Trägerinnen ohne eigene Symptome. Hämophilie A ist ein typisches Beispiel.
| Vererbungsmuster | Beispielerkrankung | Erkrankungsrisiko für Kinder |
|---|---|---|
| Autosomal-dominant | Chorea Huntington | 50 % bei einem betroffenen Elternteil |
| Autosomal-rezessiv | Mukoviszidose | 25 % bei zwei Anlageträgern |
| X-chromosomal rezessiv | Hämophilie A | 50 % der Söhne bei Trägerinnen |
Profi-Tipp: Wer eine Familiengeschichte mit unklaren neurologischen oder Stoffwechselerkrankungen hat, sollte frühzeitig eine humangenetische Beratung in Anspruch nehmen. Ein Stammbaum über drei Generationen hilft dem Genetiker, das Vererbungsmuster schnell einzugrenzen.
Welche Symptome können monogene Erkrankungen verursachen?
Monogene Erkrankungen erzeugen eine außerordentliche Vielfalt an Krankheitsbildern, weil jedes Gen in einem anderen Organ oder Zelltyp aktiv ist. Ein defektes Gen im Bindegewebe führt zu anderen Symptomen als ein defektes Gen im Nervensystem oder im Stoffwechsel. Diese Vielfalt macht die klinische Einordnung schwierig.
Ein besonders lehrreiches Beispiel ist das Ehlers-Danlos-Syndrom. Es gibt mehr als 13 genetisch verschiedene Typen, die sich alle durch überdehnbare Haut und Gelenkinstabilität äußern können, aber auf Mutationen in ganz unterschiedlichen Genen beruhen. Wer nur die Symptome kennt, kann den genauen Typ nicht bestimmen.
Genetische Heterogenität ist dabei ein zentrales Problem: Gleiche klinische Symptome können von Mutationen in verschiedenen Genen stammen. Das bedeutet, dass zwei Patienten mit identischen Beschwerden völlig unterschiedliche genetische Ursachen haben können und damit auch unterschiedlich auf Therapien ansprechen. Ohne molekulargenetische Diagnose bleibt die Behandlung oft ein Raten.
Typische Krankheitsgruppen mit monogenem Ursprung sind:
- Genetische Stoffwechselerkrankungen: Phenylketonurie (PKU), Galaktosämie, Ahornsirupkrankheit. Bei PKU fehlt ein Enzym, das die Aminosäure Phenylalanin abbaut. Ohne Behandlung entstehen schwere neurologische Schäden.
- Neuromuskuläre Erkrankungen: Spinale Muskelatrophie (SMA), Duchenne-Muskeldystrophie. Beide betreffen Muskeln und Nerven und verlaufen ohne Therapie schwer.
- Erkrankungen des Bindegewebes: Marfan-Syndrom, verschiedene Typen des Ehlers-Danlos-Syndroms.
- Erbliche Netzhauterkrankungen: Retinitis pigmentosa, Leber-kongenitale Amaurose. Diese führen zu fortschreitendem Sehverlust.
Patienten mit gleichen klinischen Symptomen können unterschiedliche genetische Ursachen haben. Das macht eine genaue molekulargenetische Diagnose nicht nur wünschenswert, sondern notwendig.
Wie erfolgt die Diagnostik monogener Erkrankungen heute?
Die molekulargenetische Diagnostik ist der Goldstandard bei Verdacht auf eine monogene Erkrankung. Drei Verfahren dominieren die klinische Praxis: die Paneldiagnostik, die Exomsequenzierung und die Genomsequenzierung. Jedes Verfahren hat seinen Platz, je nachdem wie klar der klinische Verdacht ist.
Die Paneldiagnostik untersucht eine definierte Gruppe von Genen, die für ein bestimmtes Krankheitsbild bekannt sind. Sie ist schnell und kosteneffizient, wenn der Verdacht klinisch gut eingegrenzt ist. Die Exomsequenzierung liest alle proteincodierenden Bereiche des Genoms und deckt damit den Großteil bekannter Krankheitsgene ab. Die Genomsequenzierung geht noch weiter und erfasst auch nicht-codierende Bereiche, was bei ungeklärten Fällen entscheidend sein kann.
Eine Studie aus 2026 zeigt, wie leistungsfähig diese Methoden sind: Bei Patienten mit erblichen Netzhautdystrophien erreichten Panel-, Exom- und Genomsequenzierung zusammen eine molekulargenetische Aufklärungsrate von 78 %. Das bedeutet, dass fast vier von fünf Patienten eine klare genetische Diagnose erhalten haben. Für Erkrankungen, bei denen früher jahrelange Odysseen die Regel waren, ist das ein erheblicher Fortschritt.
Profi-Tipp: Viele Patienten mit monogenen Erkrankungen erhalten zunächst eine falsche Diagnose, weil die Symptome häufigen Erkrankungen ähneln. Wer trotz Behandlung keine Besserung erlebt oder eine ungewöhnliche Familiengeschichte hat, sollte aktiv nach einem spezialisierten humangenetischen Zentrum fragen. Frühe Symptome im Kindesalter unter 6 Jahren sollten umgehend genetisch abgeklärt werden.
Die Diagnose öffnet auch den Zugang zu klinischen Studien. Viele Studien setzen eine bestätigte genetische Diagnose voraus, bevor ein Patient aufgenommen werden kann. Wer keine Diagnose hat, bleibt von diesen Möglichkeiten ausgeschlossen.
Welche Behandlungsmöglichkeiten gibt es für monogene Erkrankungen?
Die Behandlung monogener Erkrankungen reicht von einfachen diätetischen Maßnahmen bis zu hochspezialisierten Gentherapien. Welche Option infrage kommt, hängt vom betroffenen Gen, dem Organ und dem Schweregrad ab.
-
Diätetische Behandlung: Bei Phenylketonurie (PKU) lässt sich die Erkrankung durch eine phenylalaninarme Ernährung gut kontrollieren. Frühzeitig begonnen, ermöglicht diese Therapie eine normale geistige Entwicklung. Das zeigt, wie wirkungsvoll eine einfache Maßnahme sein kann, wenn die Diagnose früh gestellt wird.
-
Enzymersatztherapie: Bei bestimmten lysosomalen Speichererkrankungen wie Morbus Gaucher oder Morbus Fabry wird das fehlende Enzym regelmäßig intravenös zugeführt. Die Therapie lindert Symptome, heilt aber nicht die genetische Ursache.
-
Antisense-Oligonukleotide (ASOs): Diese synthetischen Moleküle greifen gezielt in die Genexpression ein. Bei Spinaler Muskelatrophie hat Nusinersen, ein ASO-Wirkstoff, die Prognose für betroffene Kinder grundlegend verändert.
-
Gentherapie: Dabei wird eine funktionierende Kopie des defekten Gens in die Zellen eingeschleust. Für SMA ist mit Onasemnogene abeparvovec bereits eine Gentherapie zugelassen. Aktuelle CRISPR-Anwendungen bei Sichelzellenanämie und β-Thalassämie werden derzeit in klinischen Studien erprobt und zeigen vielversprechende Ergebnisse.
-
Symptomatische Therapie: Für viele monogene Erkrankungen gibt es noch keine kausale Behandlung. Physiotherapie, Schmerzmanagement und psychosoziale Unterstützung bleiben dann die wichtigsten Säulen der Versorgung.
Die Wahl des richtigen Gentherapievektors ist dabei eine eigene wissenschaftliche Disziplin, die stark vom betroffenen Gewebe abhängt. Nicht jede Gentherapie passt zu jeder Erkrankung.
Welche Bedeutung hat die Diagnose für Betroffene und ihre Familien?
Eine genaue molekulargenetische Diagnose verändert die Situation einer Familie grundlegend. Sie ersetzt Unsicherheit durch konkretes Wissen und eröffnet neue Wege in Therapie und Beratung. Monogene Diagnosen ermöglichen präzise Risikoabschätzungen für alle Familienmitglieder.
Die Auswirkungen einer Diagnose sind vielfältig:
- Familienplanung: Eltern können das Wiederholungsrisiko für weitere Schwangerschaften einschätzen. Bei autosomal-rezessiven Erkrankungen besteht die Möglichkeit einer Präimplantationsdiagnostik.
- Zugang zu Therapien: Viele spezialisierte Behandlungen und klinische Studien setzen eine bestätigte genetische Diagnose voraus. Ohne Diagnose bleibt dieser Zugang versperrt.
- Psychosoziale Entlastung: Viele Familien berichten, dass die Diagnose trotz aller Schwere eine Erleichterung war. Das jahrelange Suchen nach einer Erklärung hat ein Ende.
- Präzisionsmedizin: Das Wissen über das exakt betroffene Protein erlaubt eine auf den Patienten zugeschnittene Behandlung, statt einer Therapie nach dem Prinzip Versuch und Irrtum.
Frühkindliche monogene Erkrankungen treten oft vor dem 6. Lebensjahr auf. Eine rechtzeitige genetische Beratung ist in diesen Fällen besonders wichtig, weil frühe Interventionen die Entwicklung des Kindes entscheidend beeinflussen können. Wer wartet, verliert Zeit, die sich später nicht zurückgewinnen lässt.
Wichtige Erkenntnisse
Monogene Erkrankungen entstehen durch eine Mutation in einem einzigen Gen und erfordern eine molekulargenetische Diagnose, um Therapiezugang, Familienplanung und präzisionsmedizinische Betreuung zu ermöglichen.
| Thema | Details |
|---|---|
| Definition Monogenerkrankung | Eine Mutation in einem Gen unterbricht die Produktion eines Proteins und löst ein charakteristisches Krankheitsbild aus. |
| Vererbungsmuster | Autosomal-dominant (50 % Risiko), autosomal-rezessiv (25 % Risiko) und X-chromosomal sind die drei Hauptmuster. |
| Diagnostik heute | Exom- und Genomsequenzierung erreichen Aufklärungsraten von bis zu 78 % bei erblichen Netzhauterkrankungen. |
| Behandlungsoptionen | Von Diät bei PKU bis zu CRISPR-basierten Gentherapien bei Sichelzellenanämie reicht das aktuelle Spektrum. |
| Bedeutung der Diagnose | Eine genaue Diagnose ermöglicht Familienplanung, Studienteilnahme und psychosoziale Entlastung. |
Meine Einschätzung: Diagnose ist keine Endstation, sondern ein Anfang
Ich erlebe immer wieder, dass Familien die Diagnose einer monogenen Erkrankung zunächst als Schock empfinden. Das ist verständlich. Aber ich halte diese Sichtweise für grundlegend falsch.
Eine molekulargenetische Diagnose ist kein Urteil. Sie ist der Moment, ab dem man wirklich handeln kann. Wer jahrelang mit unklaren Symptomen kämpft, weiß, wie zermürbend diese Ungewissheit ist. Die Diagnose beendet diese Phase. Sie gibt Familien die Sprache, mit der sie mit Ärzten, Forschern und Versicherungen sprechen können.
Was mich an der aktuellen Entwicklung begeistert: Die Lücke zwischen Diagnose und Therapie wird kleiner. Noch vor zehn Jahren war eine genetische Diagnose bei vielen seltenen Erkrankungen ein Wissen ohne Konsequenz. Heute gibt es für immer mehr monogene Erkrankungen zielgerichtete Therapien, klinische Studien oder zumindest spezialisierte Versorgungszentren. CRISPR-basierte Ansätze bei Sichelzellenanämie sind kein Zukunftsszenario mehr, sondern klinische Realität.
Mein Rat ist klar: Wer den Verdacht auf eine monogene Erkrankung hat, sollte nicht auf eine Überweisung warten, sondern aktiv ein humangenetisches Zentrum aufsuchen. Die Diagnostik seltener Erkrankungen hat sich in den letzten Jahren so stark verbessert, dass Antworten heute oft schneller kommen als erwartet. Und Antworten sind der erste Schritt zu Lösungen.
— John
Hopeatrarelabs: Spezialisierte Unterstützung bei seltenen genetischen Erkrankungen
Wer mit einer monogenen Erkrankung konfrontiert ist, steht vor Fragen, die Standardmedizin oft nicht beantworten kann. Hopeatrarelabs hat sich genau auf diese Situation spezialisiert.
Hopeatrarelabs entwickelt patientenspezifische Krankheitsmodelle aus den eigenen Zellen der Betroffenen, unter Einsatz von induzierten pluripotenten Stammzellen (iPSCs) und CRISPR-Genomeditierung. Parallel dazu werden tausende von FDA-zugelassenen Wirkstoffen, maßgeschneiderte Antisense-Oligonukleotide und Gentherapieoptionen getestet. Ziel ist es, für Erkrankungen ohne zugelassene Therapie einen möglichen Behandlungsweg zu finden. Wer mehr über seltene genetische Erkrankungen erfahren möchte, findet auf der Wissensplattform von Hopeatrarelabs umfassende Ressourcen zu Diagnose, Forschung und Therapieentwicklung.
FAQ
Was ist der Unterschied zwischen monogen und polygen?
Bei einer monogenen Erkrankung reicht eine Mutation in einem einzigen Gen aus, um die Krankheit auszulösen. Bei polygenen oder multifaktoriellen Erkrankungen wirken viele Gene zusammen, oft in Kombination mit Umweltfaktoren.
Wie hoch ist das Vererbungsrisiko bei monogenen Erkrankungen?
Das Risiko hängt vom Vererbungsmuster ab. Bei autosomal-dominanten Erkrankungen wie Chorea Huntington liegt es bei 50 %, bei autosomal-rezessiven Erkrankungen wie Mukoviszidose bei 25 %, wenn beide Elternteile Anlageträger sind.
Welche Diagnoseverfahren werden bei monogenen Erkrankungen eingesetzt?
Paneldiagnostik, Exomsequenzierung und Genomsequenzierung sind die wichtigsten Methoden. Eine Studie aus 2026 zeigte eine Aufklärungsrate von 78 % bei erblichen Netzhautdystrophien durch diese Verfahren.
Sind monogene Erkrankungen heilbar?
Die meisten monogenen Erkrankungen sind derzeit nicht vollständig heilbar, aber behandelbar. Phenylketonurie lässt sich durch Diät gut kontrollieren, und für Erkrankungen wie Spinale Muskelatrophie gibt es bereits zugelassene Gentherapien.
Ab welchem Alter sollte man eine genetische Abklärung in Betracht ziehen?
Frühe Symptome, besonders im Kindesalter unter 6 Jahren, sollten umgehend genetisch abgeklärt werden. Eine frühzeitige Diagnose verbessert die Behandlungsmöglichkeiten und die langfristige Entwicklung erheblich.