
Регуляторные Т-клетки (Treg) — это специализированная субпопуляция CD4+ лимфоцитов, которая поддерживает иммунологическую толерантность и предотвращает аутоиммунные реакции. Роль регуляторных Т-клеток при генетических болезнях определяется их способностью подавлять эффекторные лимфоциты через цитокины, прямой клеточный контакт и модуляцию дендритных клеток. Ключевой транскрипционный фактор Treg — FOXP3, мутации которого вызывают синдром IPEX: тяжелое полиорганное аутоиммунное расстройство с дебютом в раннем детстве. Гены IL2RA и MSN также критичны для численности и функции Treg, а их дефекты приводят к врожденным нарушениям иммунной системы с широким спектром клинических проявлений. Понимание этих механизмов открывает путь к генно-инженерным терапиям, способным восстановить регуляторную функцию у конкретного пациента.
Какие генетические мутации влияют на функцию регуляторных т-клеток?
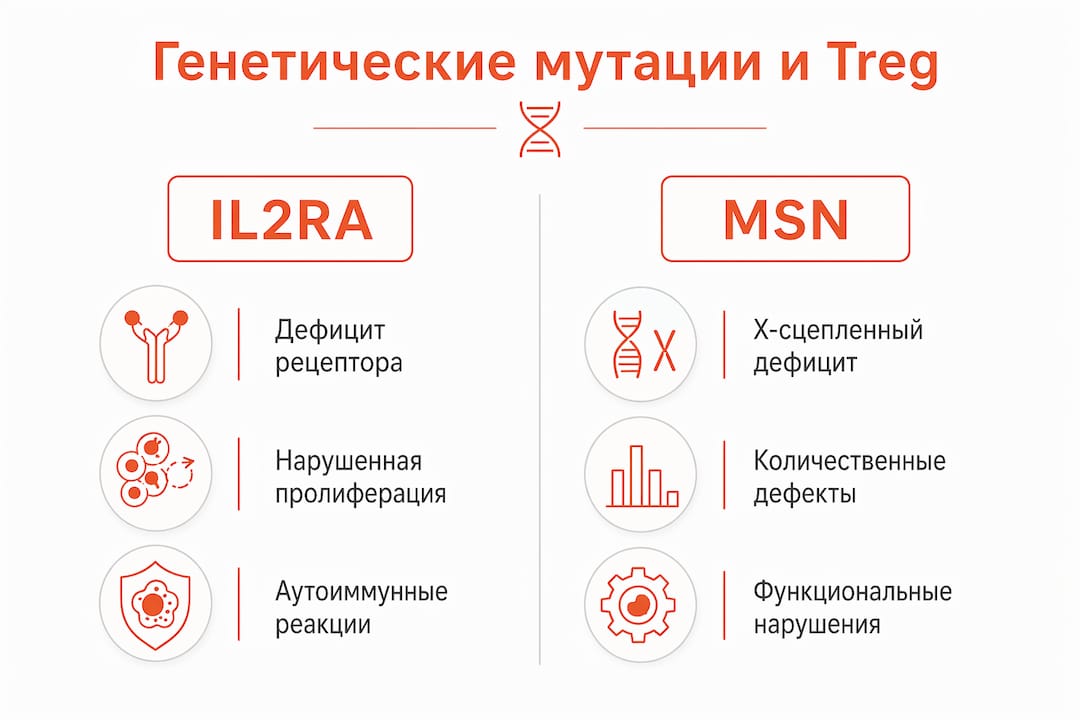
Мутации в генах IL2RA и MSN вызывают количественные и функциональные дефекты Treg, что приводит к тяжелым аутоиммунным и воспалительным заболеваниям. Это не просто снижение числа клеток: нарушается сама способность Treg распознавать сигналы активации и поддерживать супрессорный фенотип.
Мутации il2ra: дефицит рецептора интерлейкина-2
Ген IL2RA кодирует альфа-цепь рецептора интерлейкина-2 (CD25), которая необходима для выживания и пролиферации Treg. При его мутации клетки не получают достаточного сигнала IL-2 и не могут поддерживать экспрессию FOXP3 на стабильном уровне. Клинически это проявляется ранним дебютом аутоиммунного энтерита, сахарного диабета 1 типа и лимфопролиферации. Генная коррекция с помощью CRISPR-Cas9 рассматривается как альтернатива трансплантации костного мозга именно потому, что восстанавливает экспрессию CD25 в самих Treg, а не заменяет весь гемопоэтический компартмент.
Мутации MSN: x-сцепленный иммунодефицит с дефицитом treg
Новая мутация в гене MSN снижает синтез белка мезина в иммунных клетках, сокращает число Treg и усиливает активацию T-хелперов. Клинические проявления у описанных пациентов включали экзему, аутоиммунный тиреоидит и синдром Кавасаки. Это X-сцепленный дефект, что объясняет преобладание тяжелых форм у мальчиков.
Ключевые клинические последствия дефектов Treg при мутациях IL2RA и MSN:
- Аутоиммунный энтерит с ранним дебютом и резистентностью к стандартной иммуносупрессии
- Эндокринопатии (тиреоидит, сахарный диабет 1 типа) как следствие утраты периферической толерантности
- Кожные проявления (экзема, псориазиформные высыпания) при нарушении регуляции Th2-ответа
- Лимфопролиферация из-за неконтролируемой активации эффекторных Т-клеток
- Синдром Кавасаки как редкое, но задокументированное проявление дефицита MSN
Переход от концепции первичных иммунодефицитов к термину «врожденные дефекты иммунной системы» расширяет диагностический охват: теперь пациенты с поздним дебютом и атипичными формами не остаются вне поля зрения клинициста.
Для правильной классификации генетических заболеваний с иммунным компонентом необходимо учитывать не только фенотип, но и молекулярный профиль дефекта Treg.
Какие механизмы лежат в основе дисфункции treg при генетических болезнях?
Фундаментальная роль Treg состоит в подавлении эффекторных лимфоцитов через несколько параллельных механизмов: секрецию IL-10 и TGF-β, прямой цитотоксический контакт через гранзим B, а также конкурентное потребление IL-2, лишающее эффекторные клетки ростового сигнала. Нарушение любого из этих путей при генетическом дефекте ведет к системному иммунному дисбалансу.
Нестабильность foxp3 как центральный механизм
FOXP3 — не просто маркер Treg, а активный транскрипционный регулятор, поддерживающий весь супрессорный транскриптом клетки. Его экспрессия зависит от эпигенетического статуса локуса TSDR (Treg-specific demethylated region): полное деметилирование этого участка обеспечивает стабильный фенотип. При генетических дефектах, затрагивающих сигнальные пути IL-2 или структурные белки цитоскелета (как в случае MSN), деметилирование TSDR нарушается, и FOXP3 экспрессируется нестабильно.
Пластичность treg: когда регулятор становится агрессором
Treg проявляют высокую пластичность: в условиях воспаления они способны утрачивать FOXP3 и перепрограммироваться в эффекторные клетки, продуцирующие IFN-γ или IL-17. Это превращает потенциальный супрессор в активный участник воспалительного каскада. Данный феномен особенно выражен при хроническом воспалении, сопровождающем многие генетические иммунодефициты.
Последовательность событий при дисфункции Treg в условиях генетического дефекта:
- Мутация нарушает экспрессию FOXP3 или сигнальный путь IL-2/CD25
- Число функциональных Treg в периферической крови снижается
- Эффекторные Т-клетки (Th1, Th2, Th17) активируются без адекватного контроля
- Хроническое воспаление создает среду, в которой оставшиеся Treg теряют стабильность
- Перепрограммированные бывшие Treg усиливают патологический ответ
Профессиональный совет: При оценке функции Treg у пациентов с генетическими иммунодефицитами недостаточно измерять только процент CD4+CD25+FOXP3+ клеток. Необходимо анализировать метилирование TSDR и функциональную супрессорную активность in vitro, поскольку клетки с нестабильным FOXP3 сохраняют маркерный фенотип, но утрачивают функцию.
Влияние воспалительной среды на Treg носит двунаправленный характер. Умеренное воспаление может стимулировать миграцию Treg в очаг, тогда как хроническое провоспалительное микроокружение с высоким уровнем TNF-α и IL-6 запускает именно перепрограммирование. Это объясняет, почему у пациентов с одним и тем же генетическим дефектом клиническая картина может существенно различаться в зависимости от инфекционного анамнеза и воспалительной нагрузки.
Как современные технологии восстанавливают функцию регуляторных т-клеток?
CRISPR-Cas9 позволяет ex vivo корректировать дефекты генов Treg и восстанавливать экспрессию IL2RA с последующим возвратом клеток пациенту. Этот подход принципиально отличается от трансплантации костного мозга: он сохраняет аутологичный репертуар Treg и не требует миелоаблативного кондиционирования.
Crispr-cas9 для коррекции treg: от концепции к протоколу
Процедура включает выделение Treg из периферической крови пациента, электропорацию рибонуклеопротеинового комплекса Cas9/sgRNA, гомологичную рекомбинацию с донорной матрицей и последующую экспансию скорректированных клеток. Ключевое преимущество: исправленные клетки несут нативный промотор IL2RA, что обеспечивает физиологическую регуляцию экспрессии. Риск инсерционного мутагенеза при этом минимален по сравнению с ретровирусными векторами.
Car-treg и tcr-treg: направленная супрессия
CAR-Treg и TCR-Treg разрабатываются для специфического распознавания антигенов, связанных с аутоиммунными заболеваниями, предлагая альтернативу системной иммуносупрессии. CAR-Treg несут химерный антигенный рецептор, направляющий клетку к конкретному тканевому антигену, тогда как TCR-Treg используют перенесенный Т-клеточный рецептор с известной специфичностью.
Сравнение ключевых терапевтических подходов к восстановлению функции Treg:
| Подход | Механизм действия | Преимущества | Ограничения |
|---|---|---|---|
| CRISPR-коррекция Treg | Исправление мутации ex vivo | Физиологическая регуляция, аутологичность | Сложность протокола, стоимость |
| CAR-Treg | Химерный рецептор к антигену-мишени | Направленная супрессия, специфичность | Риск потери FOXP3, нестабильность |
| TCR-Treg | Перенесенный Т-клеточный рецептор | Физиологическое распознавание антигена | Ограниченный репертуар мишеней |
| Поликлональная экспансия Treg | Неспецифическое увеличение числа Treg | Простота, клинический опыт | Системная супрессия, пластичность |
Поликлональные экспансированные Treg уже прошли ранние клинические испытания: экспериментальные терапии показали эффективность при сахарном диабете 1 типа у детей. Это подтверждает принципиальную осуществимость клеточной терапии Treg, хотя вопрос долгосрочной стабильности клеток остается открытым.
Профессиональный совет: При разработке протоколов CAR-Treg не переносите напрямую условия культивирования из онкологических CAR-T программ. Treg требуют более мягкой стимуляции, специфических цитокиновых коктейлей с IL-2 низкой концентрации и регулярного контроля метилирования TSDR на каждом этапе экспансии.
Среди трудностей разработки терапии при редких генетических болезнях особое место занимает малый размер популяции пациентов, что затрудняет проведение рандомизированных испытаний для любого из перечисленных подходов.
Каковы перспективы и ограничения treg-терапии при генетических заболеваниях?
Дисфункция Treg объединяет аутоиммунные и аллергические болезни с генетической предрасположенностью в единую патогенетическую концепцию. Это открывает возможность разработки универсальных платформ терапии, адаптируемых под конкретный генетический дефект.
Реальные ограничения, с которыми сталкиваются исследователи и клиницисты сегодня:
- Пластичность после экспансии. Treg, размноженные ex vivo, могут утрачивать FOXP3 при возврате в провоспалительную среду пациента. Контроль этого процесса требует строгого менеджмента протоколов активации и цитокинового окружения.
- Принципиальное отличие CAR-Treg от CAR-T. Прямое масштабирование онкотерапии на Treg неэффективно и рискованно: Treg нуждаются в устойчивой локальной супрессии без истощения, тогда как онкологические CAR-T оптимизированы для максимальной цитотоксической активности.
- Малый размер когорт. Большинство генетических иммунодефицитов с дефектами Treg относятся к ультраредким заболеваниям, что делает классические рандомизированные испытания практически невозможными.
- Мониторинг in vivo. После введения скорректированных или инженерных Treg нет стандартизированных методов отслеживания их судьбы, локализации и функциональной активности в организме пациента.
- Регуляторные барьеры. Генно-клеточные продукты на основе Treg проходят по наиболее строгим регуляторным путям, что удлиняет путь от лаборатории до клиники.
Перспективные направления исследований сосредоточены на нескольких точках. Первое: разработка синтетических генетических цепей, поддерживающих экспрессию FOXP3 независимо от воспалительного контекста. Второе: создание «универсальных» аллогенных Treg с редактированием HLA для снижения иммуногенности. Третье: применение iPSC-технологий для получения неограниченного числа Treg с заданным генотипом. Именно iPSC-платформы, которые использует Hopeatrarelabs, позволяют моделировать дефекты Treg на клеточном уровне и тестировать корригирующие подходы до перехода к терапевтическим протоколам.
Ключевые выводы
Роль регуляторных Т-клеток при генетических болезнях определяется их способностью поддерживать иммунологическую толерантность, а мутации в генах FOXP3, IL2RA и MSN разрушают эту функцию, открывая путь к направленной генно-клеточной коррекции.
| Пункт | Подробности |
|---|---|
| Генетические мутации и Treg | Мутации IL2RA и MSN снижают число и функцию Treg, вызывая аутоиммунные и воспалительные заболевания. |
| Нестабильность FOXP3 | Воспалительная среда перепрограммирует Treg в эффекторные клетки, усиливая патологию вместо её подавления. |
| CRISPR-коррекция | Ex vivo исправление гена IL2RA восстанавливает функцию Treg и рассматривается как альтернатива трансплантации костного мозга. |
| CAR-Treg против CAR-T | Протоколы онкологической CAR-T терапии неприменимы к Treg без адаптации под требования локальной супрессии. |
| Клинические перспективы | Поликлональные Treg показали эффективность при диабете 1 типа у детей, подтверждая реализуемость клеточной терапии. |
Почему я считаю, что пластичность treg недооценивают в клинической практике
За годы работы с пациентами, у которых диагностированы генетические иммунодефициты, я убедился в одном: большинство клиницистов воспринимают Treg как статичную популяцию с фиксированной функцией. Это ошибка, которая стоит пациентам времени и здоровья.
Реальность такова, что Treg — динамичная система. Клетка, которая сегодня подавляет воспаление, завтра при смене цитокинового окружения может его усиливать. Я видел случаи, когда у пациентов с мутацией IL2RA процент FOXP3+ клеток в крови оставался в пределах нормы, но функциональная супрессорная активность была полностью утрачена. Стандартный иммунофенотип не показал бы проблемы.
Второй недооцененный момент: мультидисциплинарный подход здесь не опция, а необходимость. Иммунолог без понимания молекулярной генетики пропустит связь между конкретной мутацией и фенотипом Treg. Генетик без клинического опыта не оценит, насколько критична временная точка терапевтического вмешательства. Только совместная работа этих специалистов с биотехнологическими платформами, способными моделировать дефект на уровне пациент-специфичных клеток, дает реальный шанс на успех.
Я убежден, что будущее терапии Treg при наследственных иммунных расстройствах лежит не в универсальных протоколах, а в персонализированных моделях, где каждый генетический дефект тестируется на собственных клетках пациента до начала лечения.
— John
Исследования редких иммунных заболеваний с Hopeatrarelabs
Hopeatrarelabs специализируется на создании пациент-специфичных моделей ультраредких генетических заболеваний, включая иммунодефициты с дефектами Treg. Платформа использует iPSC-технологии и CRISPR-редактирование для воспроизведения конкретного генетического дефекта в клеточной модели, что позволяет тестировать корригирующие подходы до клинического применения.
Если вы исследуете патогенез генетических иммунодефицитов или разрабатываете терапевтические стратегии для пациентов с дефектами Treg, база знаний Hopeatrarelabs предоставляет доступ к актуальным протоколам, данным скрининга и экспертной поддержке. Для углубленного изучения возможностей точной медицины редких заболеваний на основе iPSC-моделирования посетите основной ресурс платформы.
Часто задаваемые вопросы
Что такое регуляторные т-клетки и зачем они нужны?
Регуляторные Т-клетки (Treg) — субпопуляция CD4+ лимфоцитов, экспрессирующих транскрипционный фактор FOXP3, которые подавляют эффекторные иммунные клетки и поддерживают толерантность к собственным тканям организма. Без них развиваются аутоиммунные и воспалительные заболевания.
Какие гены чаще всего нарушают функцию treg?
Мутации в генах FOXP3, IL2RA и MSN наиболее изучены как причины количественных и функциональных дефектов Treg. Дефекты FOXP3 вызывают синдром IPEX, тогда как мутации IL2RA и MSN приводят к более вариабельным клиническим фенотипам с аутоиммунными и воспалительными проявлениями.
Чем car-treg отличаются от стандартных car-t клеток?
CAR-Treg направлены на локальную супрессию иммунного ответа в конкретной ткани, тогда как онкологические CAR-T оптимизированы для максимального цитотоксического уничтожения клеток-мишеней. Прямой перенос протоколов CAR-T на Treg нарушает стабильность FOXP3 и снижает терапевтический эффект.
Можно ли применять CRISPR для лечения дефектов treg?
CRISPR-Cas9 позволяет ex vivo корректировать мутации в генах Treg, восстанавливая экспрессию IL2RA и функциональные свойства клеток. Этот подход рассматривается как альтернатива трансплантации костного мозга при врожденных иммунодефицитах с дефектами Treg.
Почему пластичность treg важна для клинициста?
Treg способны утрачивать FOXP3 в провоспалительной среде и перепрограммироваться в эффекторные клетки, усугубляя воспаление. Это означает, что стандартный иммунофенотип по маркеру FOXP3 не отражает реальную функциональную активность клеток и требует дополнительного функционального тестирования.