
Una enfermedad genética rara es una patología infrecuente cuya causa principal reside en mutaciones en uno o varios genes, y que afecta a una proporción muy pequeña de la población. El término combina dos criterios distintos: la frecuencia epidemiológica y el origen molecular. Organismos como el NIH, la Unión Europea y la Comunidad de Madrid reconocen miles de estas enfermedades, aunque la mayoría carece de tratamiento aprobado. Entender qué es una enfermedad genética rara resulta clave para pacientes, familias y estudiantes de biomedicina que buscan orientarse en un campo médico complejo y, con frecuencia, poco conocido.
¿Qué es una enfermedad genética rara y cómo se define?
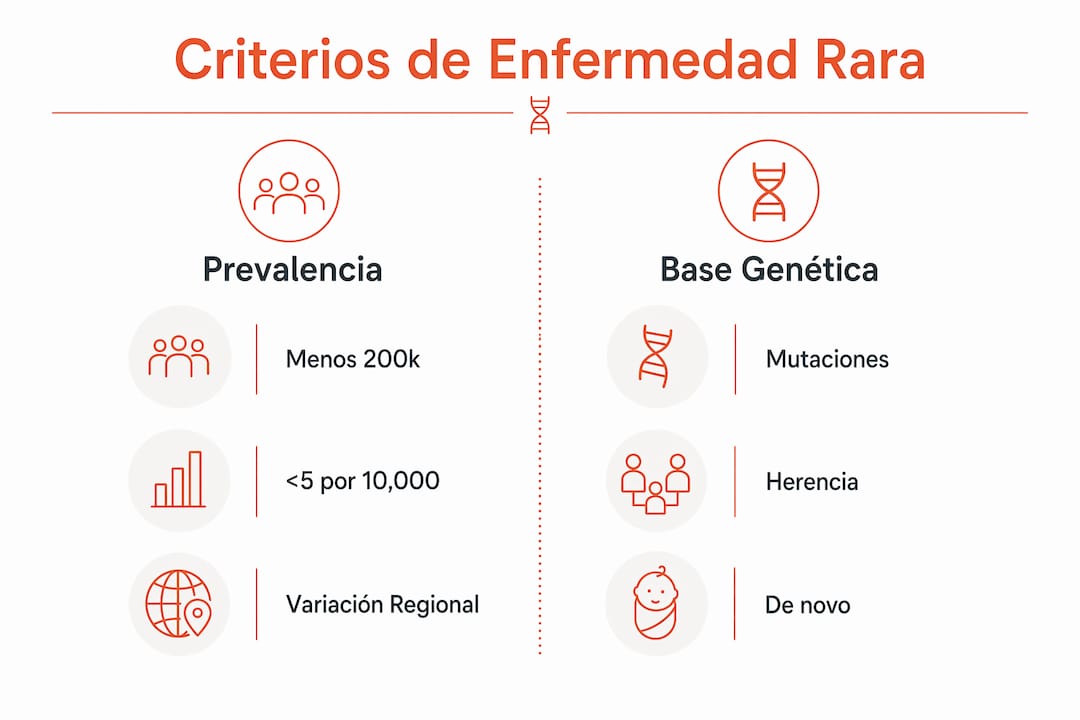
Una enfermedad genética rara reúne dos condiciones simultáneas: tiene una base genética demostrable y afecta a muy pocas personas. La base genética implica que una mutación en uno o más genes altera el funcionamiento normal de células, tejidos u órganos. La rareza, por su parte, se mide por la prevalencia en la población general.
Una fracción considerable de enfermedades raras tiene como causa mutaciones en uno o más genes, muchas transmitidas hereditariamente. Esto significa que el riesgo puede pasar de padres a hijos, aunque también existen casos de mutaciones nuevas, llamadas de novo, que no provienen de ninguno de los progenitores.
El término "rara" no implica que la enfermedad sea simple o poco grave. La baja prevalencia no implica simplicidad clínica; la complejidad diagnóstica y el manejo son altos y requieren experiencia especializada. Muchas de estas enfermedades son crónicas, progresivas y afectan gravemente la calidad de vida desde la infancia.
¿Cuáles son los criterios para considerar una enfermedad como genética y rara?
Los umbrales de prevalencia varían según el país, y esa diferencia tiene consecuencias directas sobre el acceso a recursos, medicamentos huérfanos y programas de diagnóstico. Conocer estos criterios ayuda a entender por qué una misma enfermedad puede estar reconocida oficialmente en un país y no en otro.
Los umbrales para definir enfermedad rara varían por país, lo que influye en las listas oficiales, el acceso a recursos y los programas diagnósticos disponibles. Esta variación no es un detalle menor: determina si un paciente puede acceder a medicamentos con financiación pública o a centros de referencia especializados.
| Región | Umbral de prevalencia |
|---|---|
| Estados Unidos | Menos de 200.000 personas afectadas en total |
| Unión Europea | Menos de 5 personas por cada 10.000 habitantes |
| España | Criterios similares a la UE; reconoce más de 6.000 enfermedades raras |
| Perú | Igual o menos de 1 persona por cada 2.500 habitantes |
La Unión Europea define enfermedad rara como aquella que afecta a menos de 5 personas por cada 10.000 habitantes. España aplica criterios equivalentes y reconoce más de 6.000 enfermedades raras descritas, aunque a nivel mundial se estima que existen entre 5.000 y 7.000 enfermedades distintas en este grupo, la mayoría de origen genético.
La diferencia entre una enfermedad rara y una enfermedad genética rara es conceptual. Toda enfermedad genética rara es también una enfermedad rara, pero no todas las enfermedades raras tienen causa genética confirmada. Algunas tienen origen infeccioso, autoinmune o desconocido.
Consejo profesional: Si usted o un familiar recibe un diagnóstico de enfermedad rara, verifique si está incluida en el registro oficial de su país. Ese reconocimiento formal abre puertas a financiación de pruebas genéticas, centros de referencia y ensayos clínicos.
¿Por qué es tan difícil obtener el diagnóstico de una enfermedad genética rara?
El diagnóstico de una enfermedad genética rara es uno de los procesos más exigentes de la medicina moderna. El tiempo para diagnóstico suele ser superior a 4 años en promedio, con un 20 % de pacientes esperando más de una década. Esa espera no es solo frustrante: durante ese tiempo la enfermedad avanza y las opciones terapéuticas se reducen.
Las razones de este retraso son múltiples:
- Baja prevalencia. Los médicos de atención primaria ven estos casos muy raramente, lo que dificulta el reconocimiento temprano de los síntomas.
- Síntomas inespecíficos. Muchas enfermedades genéticas raras debutan con fatiga, retraso del desarrollo o dolor crónico, síntomas comunes a decenas de condiciones más frecuentes.
- Necesidad de equipos multidisciplinares. Un solo especialista rara vez tiene toda la información necesaria. Se requiere la colaboración entre genetistas, neurólogos, pediatras y otros especialistas.
- Pruebas genéticas escalonadas. El diagnóstico genético avanza por fases: primero paneles genéticos dirigidos, luego secuenciación del exoma completo y, en casos no resueltos, tecnologías más avanzadas.
SpainUDP usa tecnologías avanzadas como el mapeo óptico del genoma y la secuenciación de lectura larga para diagnosticar enfermedades ultra-raras no identificadas con métodos tradicionales. Estas tecnologías detectan variantes estructurales complejas que los paneles estándar no pueden ver, lo que aumenta significativamente la tasa de diagnóstico definitivo en casos difíciles.
El fenotipo detallado, es decir, la descripción precisa de todos los síntomas y hallazgos clínicos, es la base de cualquier estrategia diagnóstica eficaz. Sin una documentación clínica rigurosa, incluso las mejores pruebas genéticas pueden no llegar a una conclusión.
Consejo profesional: Organice todos los informes médicos, resultados de pruebas y descripciones de síntomas en un único documento cronológico antes de cada consulta especializada. Una guía para organizar registros médicos puede marcar la diferencia entre años de espera y un diagnóstico ágil.
¿Qué ejemplos y características tienen las enfermedades genéticas raras?
Las enfermedades genéticas raras forman un grupo extraordinariamente diverso. Muchas se caracterizan por presentar síntomas crónicos, discapacitantes y progresivos que afectan la calidad y la expectativa de vida. Tres ejemplos clásicos ilustran bien esta diversidad.
La fibrosis quística es una enfermedad autosómica recesiva causada por mutaciones en el gen CFTR. Afecta principalmente a los pulmones y al páncreas, y genera una acumulación de moco espeso que deteriora la función respiratoria de forma progresiva. La enfermedad de Huntington sigue un patrón autosómico dominante: basta con heredar una copia del gen alterado para desarrollar la enfermedad, que destruye neuronas del cerebro y provoca deterioro motor y cognitivo irreversible. Las distrofias musculares, como la distrofia de Duchenne, afectan principalmente a varones y causan debilidad muscular progresiva desde la infancia.
| Tipo de enfermedad genética rara | Ejemplos | Síntomas principales |
|---|---|---|
| Autosómica recesiva | Fibrosis quística, fenilcetonuria | Problemas respiratorios, metabólicos |
| Autosómica dominante | Enfermedad de Huntington, síndrome de Marfan | Deterioro neurológico, alteraciones del tejido conectivo |
| Ligada al cromosoma X | Distrofia de Duchenne, hemofilia A | Debilidad muscular, trastornos de coagulación |
| Cromosómica | Síndrome de Down, síndrome de Turner | Discapacidad intelectual, alteraciones del desarrollo |
Más allá de los ejemplos concretos, estas enfermedades comparten rasgos estructurales. La mayoría debuta en la infancia, aunque algunas aparecen en la edad adulta. Casi todas requieren un abordaje multidisciplinar: no existe un solo especialista capaz de gestionar todos los aspectos de una enfermedad que puede afectar simultáneamente al sistema nervioso, el corazón, los riñones y el metabolismo.
¿Qué herramientas y recursos aceleran el diagnóstico y manejo de estas enfermedades?
La tecnología ha cambiado el panorama del diagnóstico en enfermedades genéticas raras durante la última década. El programa acelRare usa inteligencia artificial para procesar datos clínicos y priorizar diagnósticos diferenciales en patologías raras en minutos. Esto reduce el tiempo que un médico tarda en identificar qué pruebas pedir y a qué especialista derivar al paciente.
La captura estructurada de síntomas y antecedentes es la base de estas herramientas computacionales. Cuando los datos clínicos están bien organizados, los algoritmos pueden comparar el perfil del paciente con miles de enfermedades raras conocidas y señalar las más probables.
Otros recursos relevantes en el ámbito internacional incluyen:
- Orphanet, la base de datos europea de referencia con información sobre más de 6.000 enfermedades raras, genes implicados y centros especializados.
- OMIM (Online Mendelian Inheritance in Man), el catálogo más completo de enfermedades genéticas y sus bases moleculares, usado por genetistas clínicos en todo el mundo.
- SpainUDP, el programa español para pacientes sin diagnóstico, que aplica tecnologías de secuenciación avanzada para resolver casos que han agotado las vías diagnósticas convencionales.
- EURORDIS, la alianza europea de pacientes con enfermedades raras, que conecta a familias, investigadores y sistemas de salud en toda Europa.
Las redes internacionales de registro de pacientes también juegan un papel decisivo. Cuando los datos de cientos o miles de pacientes con la misma mutación se comparten globalmente, los investigadores pueden identificar patrones clínicos, diseñar ensayos clínicos y desarrollar tratamientos con mayor rapidez. La colaboración entre centros es, en muchos casos, la única vía para avanzar en enfermedades con decenas o pocos cientos de pacientes en todo el mundo. La medicina de precisión genética representa el siguiente paso lógico en este proceso.
Puntos clave
Las enfermedades genéticas raras combinan baja prevalencia y origen molecular, lo que exige criterios diagnósticos específicos, tecnologías avanzadas y colaboración internacional para identificarlas y tratarlas.
| Punto | Detalles |
|---|---|
| Definición combinada | Una enfermedad genética rara reúne causa genética y prevalencia inferior a los umbrales oficiales por región. |
| Umbrales variables | EE.UU. usa menos de 200.000 afectados; la UE y España aplican menos de 5 por cada 10.000 habitantes. |
| Diagnóstico tardío | El tiempo medio para obtener diagnóstico supera los 4 años; un 20 % de pacientes espera más de una década. |
| Tecnologías clave | SpainUDP y acelRare usan secuenciación avanzada e inteligencia artificial para resolver casos no diagnosticados. |
| Diversidad clínica | Fibrosis quística, Huntington y distrofias musculares ilustran la variedad de mecanismos y órganos afectados. |
Lo que nadie te dice sobre estas enfermedades
Llevo años trabajando con pacientes y familias afectadas por enfermedades genéticas raras, y hay algo que los artículos médicos rara vez reconocen: el diagnóstico no es el final del problema. Es, en muchos casos, solo el comienzo de una segunda batalla.
La medicina convencional está diseñada para enfermedades frecuentes. Cuando un paciente tiene una mutación que afecta a 300 personas en todo el mundo, no existe un protocolo estándar, no hay una guía clínica consolidada y el médico de cabecera probablemente nunca ha visto otro caso. Esa soledad clínica es real y tiene consecuencias.
Lo que me parece más esperanzador hoy es la velocidad a la que cambia el escenario tecnológico. Hace diez años, secuenciar el genoma completo de un paciente costaba decenas de miles de euros y tardaba meses. Ahora es accesible en semanas y a un coste radicalmente menor. Herramientas como SpainUDP o acelRare no son ciencia ficción: están activas, funcionan y están resolviendo casos que llevaban años sin respuesta.
Mi recomendación para cualquier familia en este proceso es concreta: no esperen a que el sistema les encuentre. Busquen activamente centros de referencia, contacten con asociaciones de pacientes y documenten cada síntoma con precisión. La información bien organizada es el recurso más valioso que un paciente puede llevar a una consulta especializada.
— John
Hopeatrarelabs: recursos especializados para enfermedades ultra-raras
Cuando el diagnóstico llega tarde o no llega, contar con un equipo especializado marca la diferencia entre años de incertidumbre y una estrategia concreta.
Hopeatrarelabs es una firma de biotecnología sanitaria centrada en enfermedades genéticas ultra-raras y casos sin diagnóstico. Su enfoque combina modelos de enfermedad personalizados a partir de las propias células del paciente, tecnologías como células madre pluripotentes inducidas (iPSCs) y edición génica con CRISPR, y cribados paralelos de miles de fármacos aprobados por la FDA. Para pacientes, familias y médicos que buscan opciones donde no las hay, el centro de conocimiento de Hopeatrarelabs ofrece recursos educativos y científicos actualizados sobre enfermedades raras, diagnóstico genético y desarrollo de terapias personalizadas.
Preguntas frecuentes
¿Cuántas enfermedades genéticas raras existen?
A nivel mundial se estiman entre 5.000 y 7.000 enfermedades raras distintas, la mayoría de origen genético. España reconoce oficialmente más de 6.000 enfermedades raras descritas.
¿Una enfermedad genética rara siempre se hereda de los padres?
No siempre. Muchas se transmiten hereditariamente, pero también existen mutaciones de novo que aparecen por primera vez en el paciente sin que ninguno de los progenitores sea portador.
¿Cómo se obtiene el diagnóstico de una enfermedad genética rara?
El diagnóstico sigue un proceso escalonado: desde paneles genéticos dirigidos hasta secuenciación del exoma completo y, en casos no resueltos, tecnologías como el mapeo óptico del genoma o la secuenciación de lectura larga empleadas por programas como SpainUDP.
¿Qué diferencia hay entre enfermedad rara y enfermedad genética rara?
Toda enfermedad genética rara es también una enfermedad rara, pero no al revés. Algunas enfermedades raras tienen origen infeccioso, autoinmune o desconocido, sin una mutación genética identificada como causa.
¿Existe tratamiento para las enfermedades genéticas raras?
La mayoría carece de tratamiento aprobado específico. Sin embargo, los avances en terapia génica, oligonucleótidos antisentido (ASOs) y cribados de fármacos existentes están abriendo opciones terapéuticas para un número creciente de estas enfermedades.