
Клиническая разработка малых молекул редких болезней — это поэтапное доказательство безопасности и эффективности низкомолекулярных соединений в строго контролируемых испытаниях на людях, начиная с получения разрешения IND и заканчивая регуляторным одобрением. В профессиональной среде этот процесс называют программой клинической разработки орфанных препаратов. Его главная сложность состоит не в химии молекулы, а в том, что пациентов катастрофически мало, естественное течение болезни плохо изучено, а стандартные статистические модели не работают. FDA и другие регуляторы ответили на этот вызов специализированными рамками, в том числе программой Rare Disease Evidence Principles (RDEP), которая меняет правила доказательства эффективности прямо сейчас.
Что такое клиническая разработка малых молекул редких болезней
Маломолекулярная терапия редких заболеваний опирается на соединения с молекулярной массой до 900 дальтон, способные проникать в клетку и воздействовать на конкретный молекулярный мишень. Это отличает их от биологических препаратов, которые действуют преимущественно на поверхностные рецепторы. Для редких генетических заболеваний такая проникающая способность критична: большинство мишеней находятся внутри клетки.
Клиническая программа начинается задолго до первого пациента. Спонсор формирует пакет доклинических данных о безопасности, фармакокинетике и механизме действия, затем подаёт заявку IND в FDA. IND — это правовой переход от лабораторных исследований к испытаниям на людях, и FDA должна либо одобрить заявку, либо выставить клинический стоп в течение 30 дней. Без активного IND ни одно клиническое исследование в США невозможно.

Редкие болезни охватывают менее 200 000 пациентов в США по определению Закона об орфанных препаратах 1983 года. Это означает, что классические рандомизированные контролируемые испытания с тысячами участников физически недостижимы. Именно поэтому FDA разработала отдельные методологические рамки, а не просто адаптировала стандартные требования.
Как адаптируются клинические испытания малых молекул к особенностям редких заболеваний
Дизайн клинических испытаний при редких болезнях строится вокруг одного принципа: максимум информации из минимального числа пациентов. Это не компромисс с качеством, а отдельная методологическая дисциплина.
Ключевые адаптации дизайна включают:
- Малые и адаптивные выборки. Исследования с 10–30 участниками становятся нормой, а не исключением. Адаптивный дизайн позволяет менять дозировку или критерии включения по ходу испытания без потери статистической валидности.
- Данные о естественном течении болезни (natural history data). Качество данных о natural history напрямую определяет регуляторное доверие к программе. Без понимания того, как болезнь прогрессирует без лечения, невозможно оценить, работает ли препарат.
- Биомаркеры как суррогатные конечные точки. Когда клинические исходы требуют лет наблюдения, валидированные биомаркеры позволяют получить сигнал об эффективности раньше.
- Стандарт ICH E8. Международный стандарт ICH E8 задаёт требования к качеству дизайна, мониторингу и управлению рисками. Малая когорта не освобождает от этих требований, а делает их соблюдение ещё более критичным.
- Функциональные исходы. Регуляторы всё чаще требуют не только биохимических маркеров, но и измеримых изменений в функциональном состоянии пациента.
Февральский проект руководства FDA 2026 года прямо указывает: спонсор должен таргетировать первопричину болезни, использовать хорошо охарактеризованные данные о её естественном течении и подтверждать механизм действия препарата. Это смещает акцент с численной статистики на механистическую доказательность.
Профессиональный совет: Начинайте сбор данных о естественном течении болезни параллельно с доклинической программой. Регуляторы оценивают качество этих данных при рассмотрении IND, и их отсутствие на старте клинической программы — одна из самых частых причин задержек.
Каковы основные этапы клинической разработки маломолекулярных препаратов при редких болезнях
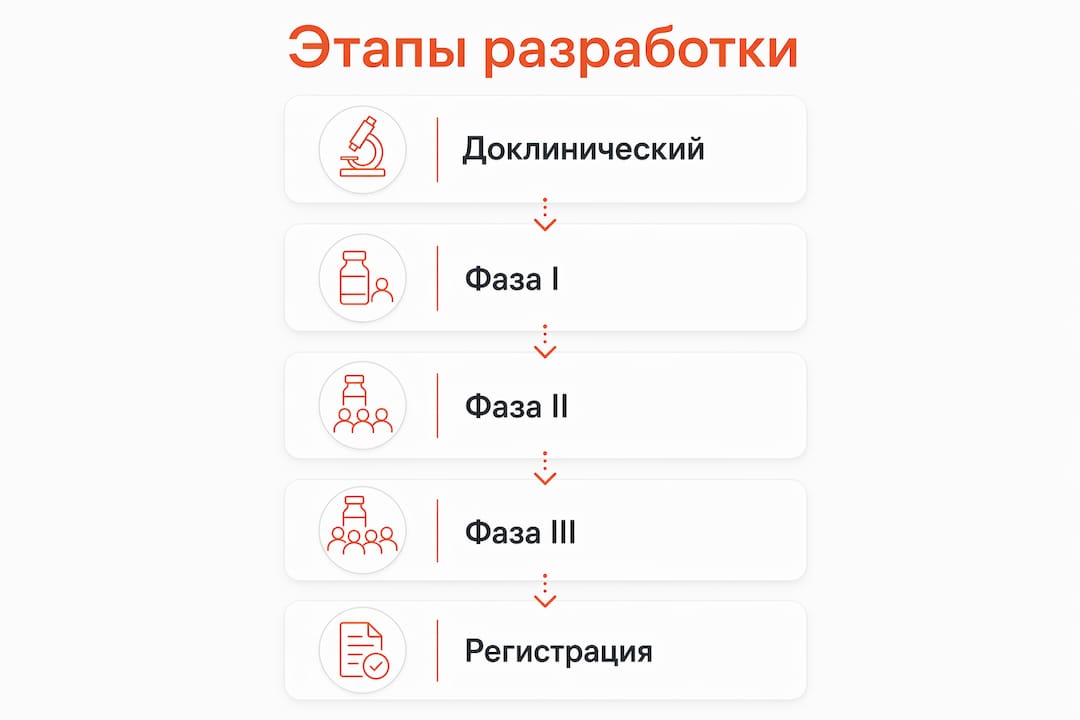
Структура клинической программы для орфанных препаратов следует общей логике фаз, но с существенными отличиями в наполнении каждого этапа.
-
Получение IND. Спонсор подаёт в FDA пакет с доклиническими данными о токсичности, фармакокинетике и предложенным клиническим протоколом. В 2026 году FDA выдала разрешение IND для UX016 — продрага сиаловой кислоты компании Ultragenyx для лечения GNE-миопатии. Старт Phase 1/2 запланирован на вторую половину 2026 года. Это наглядный пример того, как выглядит переход от доклиники к клинике в реальном времени.
-
Фаза 1 / 1b. Основная цель — безопасность, переносимость и фармакокинетический профиль. При редких болезнях фазу 1 часто объединяют с фазой 2, чтобы не тратить ограниченный пул пациентов на отдельные этапы.
-
Фаза 1b/2. Здесь оценивают предварительную эффективность, подбирают дозу и собирают данные по биомаркерам. Actio Biosciences запустила исследование KYRON Phase 1b/2 для ABS-1230 при KCNT1-эпилепсии. ABS-1230 принят в программу RDEP, что означает более тесное взаимодействие с FDA и возможность использовать гибкие стандарты доказательности.
-
Фаза 2/3 или объединённая фаза. При ультраредких болезнях фазы 2 и 3 нередко объединяют в одно исследование с заранее определёнными промежуточными и финальными конечными точками.
-
Подача NDA/BLA и регуляторное одобрение. Спонсор формирует полный пакет доказательств и подаёт заявку на одобрение. Для орфанных препаратов доступны ускоренные пути: Breakthrough Therapy, Accelerated Approval и Priority Review.
| Этап | Стандартная программа | Программа при редких болезнях |
|---|---|---|
| Размер выборки | Сотни и тысячи пациентов | 10–50 пациентов |
| Дизайн | Классический РКИ | Адаптивный, корзинный, n-of-1 |
| Конечные точки | Клинические исходы | Биомаркеры + функциональные исходы |
| Регуляторный путь | Стандартный NDA | Orphan Drug, RDEP, Breakthrough |
| Длительность наблюдения | Определяется статистикой | До 48 недель и более |
Какие особенности фармакокинетики и клинических исходов критичны для малых молекул при редких болезнях
Фармакокинетика малых молекул при редких болезнях решает задачу, которую не решает ни одна другая дисциплина: как доказать, что препарат делает то, что должен, когда у вас 15 пациентов и 48 недель наблюдения.
Оценка соотношения экспозиция/ответ (exposure-response) на малых когортах требует нестандартных подходов. Популяционный фармакокинетический анализ (PopPK) позволяет строить модели на данных от единичных пациентов и экстраполировать их на популяцию. Без PopPK подбор дозы при редких болезнях превращается в угадывание.
Критические аспекты фармакокинетического анализа при редких болезнях:
- Длительность наблюдения. Программы Phase 1/2 предусматривают сбор данных PK и функциональных исходов до 48 недель и более. Это необходимо для оценки долгосрочной безопасности и накопления доказательств эффективности.
- Цепочка биомаркеров. Формируется доказательная цепочка: механизм действия → фармакодинамика → биомаркер → клиническая польза. Каждое звено должно быть подтверждено данными, иначе регулятор не примет суррогатную конечную точку.
- Педиатрические популяции. Многие редкие болезни манифестируют в детском возрасте. Исследование KYRON для ABS-1230 охватывает пациентов до 21 года, что требует отдельного педиатрического PK-анализа и специфических критериев безопасности.
- Функциональные исходы. Регуляторы ожидают измеримых изменений в качестве жизни или функциональном статусе пациента, а не только биохимических сдвигов.
Профессиональный совет: Встраивайте PopPK-анализ в протокол с первого дня, а не добавляйте его постфактум. FDA всё чаще запрашивает PopPK-модели как часть пакета доказательств уже на этапе Phase 1b/2, особенно для педиатрических популяций.
Выбор конечных точек при редких болезнях — отдельная стратегическая задача. Клинически значимые исходы могут требовать лет наблюдения, которых у программы нет. Валидированный биомаркер решает эту проблему, но только если его связь с клинической пользой доказана. Именно поэтому понимание того, почему нет одобренных лечений для многих редких болезней, начинается с провала на этапе выбора конечных точек.
Какие регуляторные инновации меняют разработку лекарств для редких заболеваний
FDA последовательно строит регуляторную архитектуру, которая признаёт: стандартные требования к доказательствам неприменимы к болезням с 50 пациентами в мире.
Ключевые регуляторные инструменты 2025–2026 годов:
- RDEP (Rare Disease Evidence Principles). Программа предполагает раннее взаимодействие спонсора с FDA для согласования нестандартных доказательных подходов. Принятие ABS-1230 в RDEP означает, что Actio Biosciences и FDA совместно определяют, какие данные будут достаточными для одобрения.
- Draft Guidance FDA февраль 2026. Документ требует от спонсоров таргетировать первопричину болезни, использовать данные о её естественном течении и подтверждать механизм действия. Это смещает стандарт доказательности от статистики к биологии.
- Ускоренные пути одобрения. Breakthrough Therapy Designation, Accelerated Approval и Priority Review доступны для орфанных препаратов и существенно сокращают время до рынка.
- Альтернативные дизайны исследований. Корзинные испытания, платформенные исследования и дизайн n-of-1 получают регуляторное признание как инструменты для ультраредких болезней.
«Переход от традиционных рандомизированных контролируемых испытаний к механизм-ориентированным доказательствам — это не снижение стандартов. Это признание того, что биологическая достоверность важнее статистической мощности, когда пациентов единицы.»
Риски новых подходов реальны. Гибкость в требованиях к доказательствам создаёт давление на спонсоров: нужно самостоятельно обосновывать каждое методологическое решение, а не следовать стандартному шаблону. Это требует более глубокой научной экспертизы на этапе планирования программы.
Ключевые выводы
Клиническая разработка маломолекулярных препаратов для редких болезней требует механистической доказательности, адаптивного дизайна и раннего взаимодействия с FDA через программы RDEP и IND.
| Пункт | Подробности |
|---|---|
| Разрешение IND | Обязательный первый шаг: без активного IND клинические испытания в США невозможны. |
| Адаптивный дизайн | Малые выборки требуют адаптивных протоколов и PopPK-анализа с первого дня программы. |
| Данные о natural history | Качество данных о естественном течении болезни определяет регуляторное доверие ко всей программе. |
| Программа RDEP | Принятие в RDEP позволяет согласовать нестандартные доказательные подходы напрямую с FDA. |
| Цепочка биомаркеров | Каждое звено от механизма действия до клинической пользы должно быть подтверждено данными. |
Что я думаю о клинической разработке малых молекул при редких болезнях
За годы работы с программами орфанных препаратов я пришёл к одному неудобному выводу: большинство задержек в клинических программах при редких болезнях происходят не из-за проблем с молекулой, а из-за недостаточной подготовки доказательной стратегии до подачи IND.
Команды тратят годы на оптимизацию химии соединения и месяцы на доклинические токсикологические пакеты. Но вопрос «какие данные убедят FDA, что это работает при 20 пациентах» часто остаётся без ответа до начала Phase 1. Это системная ошибка, и она дорого стоит.
Программа RDEP меняет ситуацию, но только для тех, кто использует её правильно. Раннее взаимодействие с FDA через RDEP — это не формальность, а возможность получить предварительное согласие на нестандартные конечные точки и дизайн. Команды, которые воспринимают RDEP как галочку в списке, упускают главное.
Ещё один недооценённый аспект: педиатрические популяции при редких болезнях требуют отдельной стратегии с самого начала. Добавить педиатрический компонент постфактум почти всегда означает переделывать протокол и терять время. Пример KYRON с охватом пациентов до 21 года показывает, что это можно и нужно закладывать в дизайн изначально.
Перспективы на ближайшие годы я оцениваю осторожно. Регуляторная гибкость растёт, но вместе с ней растут и ожидания по качеству научного обоснования. Спонсоры, которые думают, что RDEP означает «меньше данных», столкнутся с разочарованием. Это означает «другие данные», и разница принципиальная.
— John
Hopeatrarelabs: ресурсы для разработчиков орфанных препаратов
Hopeatrarelabs создан для исследователей, которые работают на переднем крае разработки терапий для ультраредких болезней. Платформа объединяет методологические материалы по дизайну клинических программ, данные о регуляторных подходах FDA и практические инструменты для планирования доказательной стратегии.
Если вы разрабатываете маломолекулярный препарат для редкого заболевания и ищете структурированную базу знаний по клиническим этапам, регуляторным требованиям и примерам реальных программ 2026 года, база знаний Hopeatrarelabs охватывает весь цикл: от IND до регуляторного одобрения. Для тех, кто только начинает разбираться в точной медицине при ультраредких болезнях, платформа предлагает введение в iPSC-моделирование и параллельный скрининг терапий.
Часто задаваемые вопросы
Что такое IND и зачем он нужен для клинических испытаний?
IND (Investigational New Drug) — это разрешение FDA на проведение испытаний нового препарата на людях. Без активного IND ни одно клиническое исследование в США не может начаться.
Чем RDEP отличается от стандартного пути одобрения орфанных препаратов?
RDEP предполагает раннее взаимодействие спонсора с FDA для согласования нестандартных доказательных подходов, включая малые выборки и механизм-ориентированные конечные точки. Это не ускоренный путь одобрения, а инструмент совместного планирования доказательной стратегии.
Почему данные о естественном течении болезни так важны при редких заболеваниях?
Без понимания того, как болезнь прогрессирует без лечения, невозможно оценить эффект препарата на малой выборке. FDA прямо требует использования хорошо охарактеризованных данных о natural history в проекте руководства 2026 года.
Как выбрать конечные точки для клинического испытания при редком заболевании?
Конечные точки строятся по цепочке: механизм действия → фармакодинамика → биомаркер → клиническая польза. Каждое звено должно быть подтверждено данными, иначе регулятор не примет суррогатную конечную точку как доказательство эффективности.
Можно ли объединять фазы клинических испытаний при редких болезнях?
Да. При ультраредких болезнях фазы 1 и 2 или фазы 2 и 3 нередко объединяют в одно исследование с заранее определёнными промежуточными и финальными конечными точками. Это позволяет не тратить ограниченный пул пациентов на отдельные этапы.