
As células-tronco pluripotentes induzidas, conhecidas como iPSCs, são definidas como células adultas reprogramadas geneticamente para um estado semelhante ao embrionário, capazes de se diferenciar em qualquer tipo celular do organismo. Na iPSC modelação doença neurológica rara, esta tecnologia permite criar neurónios com as mutações exatas do paciente, reproduzindo a doença em laboratório com uma fidelidade impossível de alcançar com modelos animais. O diagnóstico de doenças neurológicas raras demora em média 5,4 anos após os primeiros sintomas. Este atraso torna urgente o desenvolvimento de ferramentas que acelerem tanto o estudo como o tratamento destas condições.
Como as iPSCs modelam doenças neurológicas raras no laboratório
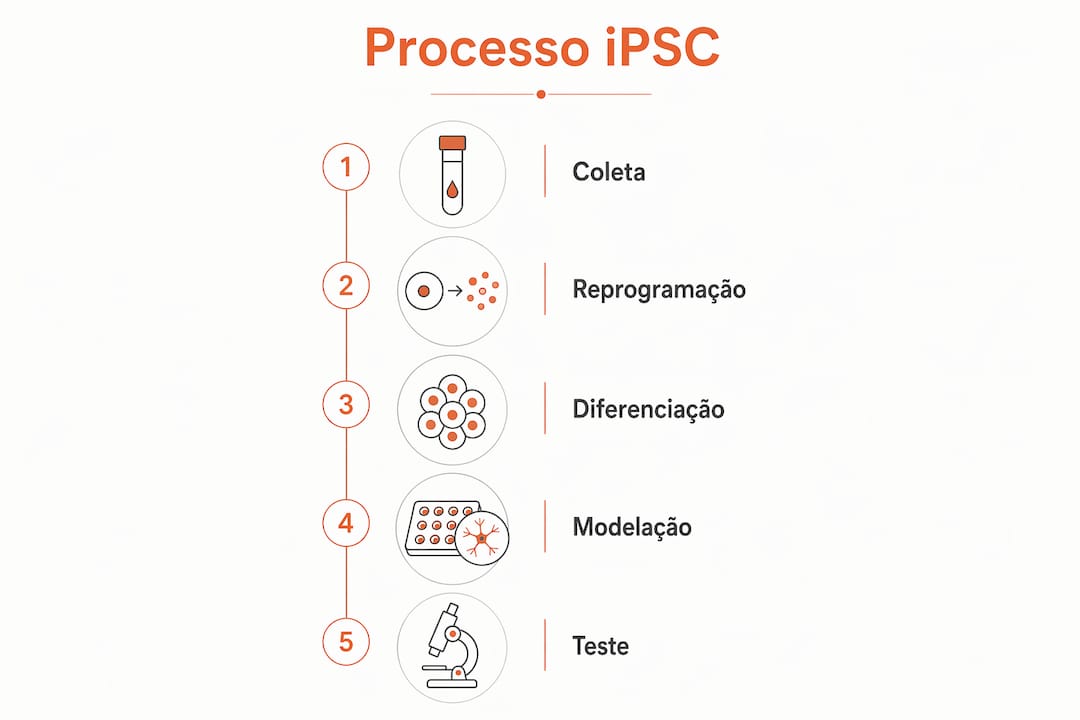
A modelação por iPSC começa com uma amostra de células do próprio paciente, geralmente células da pele ou do sangue. Essas células são reprogramadas em laboratório através da introdução de fatores de transcrição específicos, como os descritos por Shinya Yamanaka, revertendo-as para um estado pluripotente. A partir daí, os investigadores diferenciam essas iPSCs em neurónios, astrócitos ou oligodendrócitos, consoante o tipo celular afetado pela doença.
O resultado é um modelo celular que carrega as mutações genéticas do paciente. Isto significa que o comportamento anormal das células, como acumulação de proteínas tóxicas ou disfunção mitocondrial, pode ser observado diretamente. Os modelos celulares de doenças raras permitem estudar patologias genéticas com uma precisão que os modelos animais não conseguem replicar, especialmente em doenças com mecanismos exclusivamente humanos.
A utilidade prática mais imediata é a triagem de fármacos. Com neurónios derivados de iPSC do paciente, é possível testar centenas ou milhares de compostos aprovados pela FDA para identificar quais corrigem o defeito celular. Esta abordagem reduz drasticamente o tempo e o custo do desenvolvimento terapêutico em doenças sem tratamento aprovado.
As vantagens face aos modelos tradicionais são claras. Os modelos animais frequentemente não reproduzem a fisiopatologia humana com precisão suficiente. As iPSCs eliminam essa barreira ao colocar a biologia do paciente no centro da investigação.
- Recolha de células do paciente (pele, sangue ou urina)
- Reprogramação para estado pluripotente mediante fatores de transcrição
- Diferenciação em tipos celulares neurais relevantes para a doença
- Caracterização do modelo para confirmar que reproduz os defeitos celulares conhecidos
- Triagem de compostos terapêuticos no modelo validado
Dica profissional: Ao avaliar um programa de modelação iPSC, pergunte sempre se o modelo foi validado contra biomarcadores celulares conhecidos da doença. Um modelo não validado pode gerar resultados enganosos na triagem de fármacos.
Doenças neurológicas raras já modeladas com iPSCs
A doença de Niemann-Pick tipo C é um dos exemplos mais documentados de iPSC modelação doença neurológica rara. Um estudo de 2026 demonstrou que neurónios derivados de pacientes com a mutação NPC1 I1061T, quando tratados com estabilizadores de proteostase, aumentaram os níveis da proteína mutante. Este resultado confirma que o modelo iPSC reproduz fielmente o defeito molecular da doença.
O mesmo modelo revelou que o tratamento com 2-hydroxypropyl-β-cyclodextrin (HPβCD) corrigiu defeitos celulares in vitro, incluindo a acumulação anormal de colesterol e a disfunção lisossomal. Este composto já está em ensaios clínicos para Niemann-Pick tipo C. A ligação direta entre o resultado no modelo iPSC e o ensaio clínico demonstra o valor translacional desta abordagem.
Na ataxia espinocerebelosa tipo 1 (SCA1), investigadores criaram linhas celulares isogénicas a partir de iPSCs de pacientes para estudar o impacto da mutação ATXN1. Estas linhas permitem comparar células com e sem a mutação num fundo genético idêntico, isolando o efeito causal da doença. Os estudos sobre iPSC nesta patologia abriram caminho para testes de compostos que estabilizam a proteína ataxina-1 mutante.
Os avanços em terapias personalizadas para doenças raras neurodegenerativas usando iPSC são reportados em diversas publicações científicas recentes. O padrão que emerge é consistente: o modelo iPSC identifica um mecanismo, sugere um alvo terapêutico e orienta a seleção de compostos para ensaio clínico.
Exemplos de doenças neurológicas raras modeladas com iPSCs:
- Doença de Niemann-Pick tipo C: modelo NPC1 I1061T validado com HPβCD e estabilizadores de proteostase
- Ataxia espinocerebelosa tipo 1: linhas isogénicas para estudo da proteína ataxina-1
- Síndrome de Rett: neurónios iPSC revelaram défices sinápticos corrigíveis com IGF-1
- Doença de Huntington: modelos iPSC identificaram vulnerabilidade específica de neurónios estriatais
- Esclerose lateral amiotrófica familiar: neurónios motores iPSC com mutações SOD1 e TDP-43 usados em triagens de fármacos
| Doença | Mutação modelada | Composto testado | Resultado |
|---|---|---|---|
| Niemann-Pick tipo C | NPC1 I1061T | HPβCD | Correção de defeitos lisossomais in vitro |
| Ataxia espinocerebelosa tipo 1 | ATXN1 CAG expandido | Compostos estabilizadores | Redução de agregados proteicos |
| Síndrome de Rett | MECP2 | IGF-1 | Melhoria de défices sinápticos |
| Doença de Huntington | HTT CAG expandido | Compostos antiagregação | Redução de toxicidade neuronal |
Como o CRISPR melhora os modelos iPSC em doenças neurológicas raras
A edição genética CRISPR é definida como uma ferramenta molecular que permite cortar e modificar sequências específicas de ADN com precisão. Aplicada à cultura de células iPSC, o CRISPR resolve um problema central nos estudos com iPSC: a variabilidade genética entre pacientes diferentes torna difícil atribuir um resultado terapêutico à mutação específica e não ao fundo genético individual.
A solução são as linhas celulares isogénicas, consideradas o padrão-ouro nos estudos com iPSC. O CRISPR corrige a mutação numa linha iPSC do paciente, criando uma linha de controlo geneticamente idêntica exceto na mutação de interesse. Qualquer diferença observada entre as duas linhas é atribuível exclusivamente à mutação. Esta abordagem elimina o ruído genético que comprometia estudos anteriores.
Benefícios concretos do CRISPR na modelação iPSC:
- Controlo genético rigoroso: linhas isogénicas eliminam variáveis de fundo genético
- Introdução de mutações específicas: permite criar modelos de doenças em iPSCs saudáveis para validação
- Correção terapêutica in vitro: testa se a correção da mutação reverte o fenótipo celular
- Validação de alvos terapêuticos: confirma que um alvo é causal antes de avançar para ensaio clínico
Na ataxia espinocerebelosa tipo 1, a geração de linhas isogénicas por CRISPR permitiu confirmar que a expansão CAG no gene ATXN1 é suficiente para causar os defeitos celulares observados. Sem este controlo, os resultados poderiam refletir diferenças genéticas entre dadores e não o efeito da mutação. O CRISPR transformou a modelação iPSC de uma ferramenta descritiva numa plataforma causal.
Dica profissional: Quando um laboratório apresenta resultados de triagem de fármacos em iPSCs, pergunte se usaram linhas isogénicas como controlo. Sem esse controlo, a eficácia de um composto pode ser um artefacto da variabilidade genética entre dadores.
Para familiares que investigam opções terapêuticas, o guia prático sobre CRISPR na modelação de doenças raras explica como esta tecnologia é aplicada em contexto clínico e de investigação.
Quais são os desafios atuais da modelação iPSC em neurologia?
A modelação por iPSC enfrenta limitações reais que qualquer família deve conhecer antes de interpretar resultados. O desafio mais discutido é a diferença entre modelos bidimensionais e tridimensionais. Os modelos 2D são mais simples e reprodutíveis, mas não capturam a arquitetura do tecido nervoso. Os organoides cerebrais, estruturas 3D que imitam o desenvolvimento do cérebro humano, revelam alterações que os modelos planos não detetam.
Estudos com organoides cerebrais que modelam mutações ligadas a epilepsia grave observaram redução precoce de células progenitoras neurais e padrões de crescimento anormais. Estes achados não eram visíveis em modelos 2D. Isto significa que a escolha do modelo condiciona diretamente o que se consegue descobrir sobre a doença.
| Dimensão | Vantagens | Limitações |
|---|---|---|
| Modelos 2D | Reprodutíveis, baratos, fáceis de escalar para triagem | Não capturam arquitetura tecidual; menos relevantes para fenótipos complexos |
| Organoides 3D | Reproduzem desenvolvimento cerebral; revelam fenótipos ausentes em 2D | Variabilidade entre preparações; difíceis de escalar; custo elevado |
A heterogeneidade genotípica e fenotípica é outro obstáculo. Duas pessoas com a mesma mutação podem ter apresentações clínicas diferentes. Um modelo iPSC captura a biologia de um paciente específico, não necessariamente de todos os pacientes com a mesma doença. Isto limita a generalização dos resultados.
O custo e a complexidade técnica da cultura de células iPSC continuam a ser barreiras para muitos centros. A diferenciação em tipos celulares neurais específicos exige protocolos longos e especializados. Contudo, os avanços em protocolos de diferenciação acelerada, como os sistemas i3Neuron, estão a reduzir o tempo de 6–8 semanas para menos de 3 semanas. O futuro da modelação iPSC passa por organoides vascularizados, co-culturas com células imunes e integração com inteligência artificial para análise de imagem celular em larga escala.
Principais conclusões
A modelação por iPSC é a abordagem mais direta para criar modelos celulares específicos do paciente em doenças neurológicas raras, acelerando a identificação de terapias onde nenhuma existe.
| Ponto | Detalhes |
|---|---|
| iPSCs reproduzem a doença do paciente | Neurónios derivados de iPSC carregam as mutações exatas, permitindo estudar mecanismos celulares reais. |
| CRISPR garante rigor científico | Linhas isogénicas criadas por CRISPR isolam o efeito da mutação e eliminam variáveis genéticas de fundo. |
| Exemplos clínicos validam a abordagem | Niemann-Pick tipo C e ataxia espinocerebelosa tipo 1 têm modelos iPSC com resultados terapêuticos documentados. |
| Organoides superam limitações dos modelos 2D | Modelos tridimensionais revelam fenótipos celulares ausentes em culturas planas, aumentando a relevância clínica. |
| Diagnóstico tardio torna a modelação urgente | Com um atraso médio de 5,4 anos no diagnóstico, ferramentas como iPSC são essenciais para acelerar o acesso a terapias. |
O que aprendi acompanhando famílias neste caminho
Ao longo dos anos, acompanhei famílias que chegaram ao laboratório com um diagnóstico recente de uma doença neurológica rara e uma pergunta simples: «existe alguma coisa que possamos fazer?». A modelação por iPSC mudou a resposta a essa pergunta de forma concreta.
O que me impressiona não é a sofisticação técnica. É a velocidade com que um modelo celular pode passar de «não sabemos o que causa este fenótipo» para «este composto corrige o defeito em neurónios do vosso filho». Esse salto, que antes demorava décadas, está a comprimir-se para meses em casos bem documentados.
Há, porém, uma expectativa que precisa de ser gerida com cuidado. Um resultado positivo num modelo iPSC não é um tratamento. É uma pista forte. A distância entre a célula numa placa de Petri e um ensaio clínico aprovado ainda é real e exige trabalho adicional. As famílias que compreendem esta distinção tomam decisões mais informadas e mantêm-se mais resilientes ao longo do processo.
O acesso a tratamentos experimentais em doenças raras está a tornar-se mais estruturado precisamente porque a modelação iPSC fornece dados pré-clínicos que justificam a abertura de protocolos compassivos. Isto é progresso real, ainda que lento para quem vive com urgência.
— John
Hopeatrarelabs: modelação iPSC para a sua doença rara
A Hopeatrarelabs é uma empresa de biotecnologia especializada em criar modelos celulares personalizados para doenças ultra-raras e não diagnosticadas, usando iPSCs e edição genética CRISPR. O processo inclui a geração de neurónios derivados das células do paciente, triagem paralela de milhares de fármacos aprovados pela FDA, avaliação de oligonucleótidos antisense (ASOs) e análise de opções de terapia génica.
Para familiares e pacientes que procuram compreender o que a ciência pode oferecer hoje, a central de recursos da Hopeatrarelabs reúne guias detalhados sobre modelação iPSC, CRISPR e acesso a terapias experimentais. A abordagem é transparente, cientificamente rigorosa e centrada na urgência de cada caso. Saiba mais sobre os serviços de medicina de precisão disponíveis para a sua situação específica.
Perguntas frequentes
O que são iPSCs e para que servem em doenças raras?
As iPSCs são células adultas reprogramadas para um estado pluripotente, capazes de se diferenciar em qualquer tipo celular, incluindo neurónios. Em doenças neurológicas raras, permitem criar modelos celulares com as mutações do paciente para estudar a doença e testar terapias.
Como a modelação por iPSC ajuda a encontrar tratamentos?
Os neurónios derivados de iPSC do paciente são usados para testar compostos terapêuticos em laboratório, identificando quais corrigem o defeito celular antes de avançar para ensaios clínicos. Esta triagem personalizada é especialmente útil em doenças sem tratamento aprovado.
O que são linhas isogénicas e por que são importantes?
Linhas isogénicas são iPSCs geneticamente idênticas exceto numa mutação específica, criadas por edição CRISPR. Garantem que os resultados observados são causados pela mutação da doença e não por diferenças genéticas entre dadores.
Quais doenças neurológicas raras já têm modelos iPSC validados?
A doença de Niemann-Pick tipo C, a ataxia espinocerebelosa tipo 1, a síndrome de Rett e a doença de Huntington têm modelos iPSC documentados com resultados terapêuticos publicados. Estes modelos já orientaram a seleção de compostos para ensaios clínicos.
Onde posso obter mais informação sobre modelação iPSC para o meu caso?
A Hopeatrarelabs disponibiliza recursos detalhados sobre modelação iPSC e opções terapêuticas personalizadas na sua central de conhecimento, com guias acessíveis para familiares e pacientes.