
Геномное импринтирование — это эпигенетический процесс, при котором активность гена определяется тем, от кого он унаследован: от матери или от отца. Это не мутация в ДНК, а особая метка, которая включает или выключает ген в зависимости от родительского происхождения. Понять, что такое геномное импринтирование редкой болезни, значит разобраться в одном из самых тонких механизмов наследственности. В человеческом геноме около 100 генов регулируются именно так. Нарушения этого процесса лежат в основе синдромов Прадера-Вилли и Ангельмана, а также ряда других редких заболеваний, диагностика которых до сих пор остаётся сложной задачей.
Что такое геномное импринтирование редкой болезни: механизмы и основы
Геномное импринтирование работает через два главных молекулярных инструмента: метилирование ДНК и модификацию гистонов. Метилирование — это присоединение химической группы к промотору гена, что блокирует его считывание. Модификация гистонов меняет плотность упаковки ДНК, делая ген недоступным для транскрипционной машины клетки. Оба процесса работают согласованно и устанавливаются ещё в зародышевых клетках родителей.
Ключевой принцип импринтинга прост: из двух копий гена (от матери и от отца) активна только одна. Какая именно, зависит от родительского происхождения. Это отличает импринтинг от обычного аутосомно-доминантного или рецессивного наследования, где обе копии потенциально активны. Если активная копия повреждена или утрачена, резервной нет.
Эволюционное объяснение этого феномена связано с конфликтом родительских генов. Отцовские гены «заинтересованы» в максимальном росте плода, тогда как материнские ограничивают его, сохраняя ресурсы матери. Импринтинг стал молекулярным компромиссом этого эволюционного противостояния. Именно поэтому многие импринтированные гены регулируют рост и развитие плода.

Комплекс поликомб (Polycomb repressive complex) участвует в поддержании репрессивных меток на протяжении всей жизни клетки. Это означает, что однажды установленный импринтинг стабильно передаётся при делении клеток. Нарушение этой стабильности ведёт к болезни.
Профессиональный совет: Если вы изучаете генетику редкого заболевания в семье, уточните у генетика, проводился ли анализ метилирования ДНК, а не только стандартное секвенирование. Эти два метода дают принципиально разную информацию.
Импринтинг также объясняет, почему одна и та же делеция на хромосоме 15 вызывает два совершенно разных синдрома. Всё зависит от того, чья копия утрачена: отцовская или материнская.
Какие редкие болезни связаны с нарушениями импринтинга?
Синдром Прадера-Вилли и синдром Ангельмана — самые изученные примеры болезней импринтинга. Потеря отцовских генов на хромосоме 15 вызывает синдром Прадера-Вилли, а потеря материнской копии гена UBE3A — синдром Ангельмана. Оба синдрома затрагивают один и тот же участок хромосомы, но клинически выглядят совершенно по-разному.
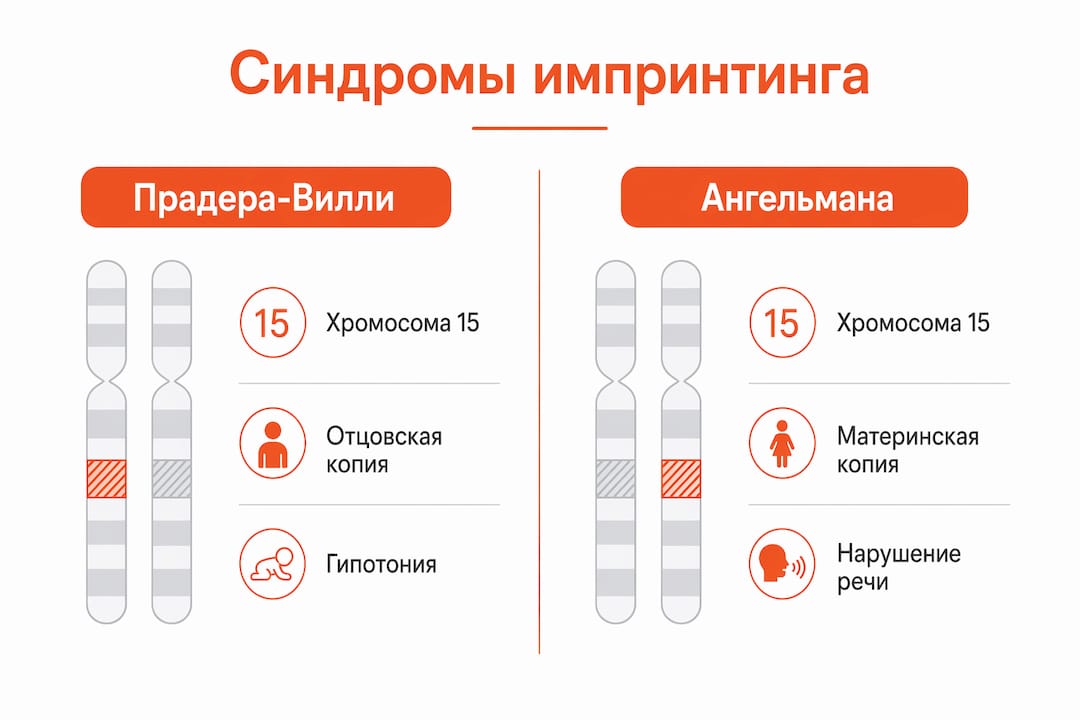
Синдром Прадера-Вилли проявляется гипотонией у новорождённых, задержкой развития, неконтролируемым аппетитом и ожирением. Синдром Ангельмана характеризуется тяжёлой умственной отсталостью, отсутствием речи, судорогами и характерным «счастливым» поведением. Оба состояния требуют пожизненной поддержки.
Помимо этих двух синдромов, нарушения импринтинга лежат в основе ряда других редких состояний:
- Синдром Беквита-Видемана — избыточный рост плода и повышенный риск опухолей, связан с нарушением импринтинга на хромосоме 11.
- Синдром Сильвера-Рассела — задержка роста и асимметрия тела, вызвана нарушением импринтинга того же региона хромосомы 11, но с противоположным эффектом.
- Псевдогипопаратиреоз типа 1b — нарушение реакции на паратгормон, связанное с импринтингом гена GNAS.
- Синдром Темпл и синдром Кавасима — редкие состояния с нарушениями роста и метаболизма.
| Синдром | Хромосома | Затронутый родитель | Основные симптомы |
|---|---|---|---|
| Прадера-Вилли | 15 | Отцовская копия | Ожирение, гипотония, задержка развития |
| Ангельмана | 15 | Материнская копия (UBE3A) | Судороги, отсутствие речи, умственная отсталость |
| Беквита-Видемана | 11 | Материнская копия | Избыточный рост, риск опухолей |
| Сильвера-Рассела | 11 | Отцовская копия | Задержка роста, асимметрия |
Около 80% редких заболеваний имеют генетическую природу. Это означает, что нарушения импринтинга составляют значимую долю среди всех генетически обусловленных редких болезней. Понимание типов редких генетических заболеваний помогает правильно выстроить диагностический путь.
Нарушения импринтинга возникают несколькими путями. Первый — делеция участка хромосомы. Второй — однородительская дисомия, когда обе копии хромосомы унаследованы от одного родителя. Третий — эпигенетический сбой в зародышевых клетках без какой-либо мутации в последовательности ДНК. Последний вариант особенно сложен для диагностики, потому что стандартное секвенирование его не выявляет.
Как диагностируют нарушения геномного импринтирования?
Диагностика болезней импринтинга требует специальных методов, которые выходят за рамки стандартного генетического анализа. Обычное секвенирование ДНК показывает последовательность нуклеотидов, но не показывает, какие гены заблокированы эпигенетическими метками. Это принципиальное ограничение.
Современный подход строится на нескольких шагах:
-
Клиническая оценка. Врач-генетик анализирует симптомы и формирует гипотезу о конкретном синдроме. Для синдрома Прадера-Вилли, например, характерна гипотония у новорождённых в сочетании с трудностями кормления.
-
Хромосомный микроматричный анализ. Выявляет делеции и дупликации на хромосомах. Обнаруживает делеции на хромосоме 15, характерные для синдромов Прадера-Вилли и Ангельмана.
-
Анализ метилирования ДНК. Определяет, какая копия гена активна. Это ключевой тест для болезней импринтинга: он выявляет как делеции, так и однородительскую дисомию, и эпигенетические сбои.
-
Полногеномное секвенирование. Сочетание полногеномного секвенирования с эпигенетическим анализом повышает точность выявления ультраредких наследственных заболеваний. Российские клиницисты уже применяют этот подход при описании клинических случаев ультраредких болезней.
-
Анализ родительского происхождения. При подозрении на однородительскую дисомию проводят генотипирование обоих родителей для подтверждения диагноза.
Главная проблема диагностики состоит в том, что патологии импринтинга не всегда связаны с мутациями в ДНК. Эпигенетические метки могут быть нарушены без изменения самой последовательности генов. Это означает, что пациент с нормальным результатом стандартного секвенирования может всё равно иметь болезнь импринтинга.
Профессиональный совет: При обращении к генетику прямо спросите: «Проводился ли анализ метилирования ДНК для исключения болезней импринтинга?» Этот вопрос помогает избежать диагностических задержек, которые нередко составляют несколько лет.
Задержка диагностики при редких болезнях остаётся серьёзной проблемой. Семьи годами ищут объяснение симптомам, проходя через множество специалистов. Понимание того, что такое редкое генетическое заболевание, помогает задавать правильные вопросы с самого начала.
Какие перспективы лечения открывает изучение импринтинга?
Терапия болезней импринтинга сталкивается с уникальными трудностями. Стандартная генная терапия направлена на замену дефектного гена, но при болезнях импринтинга проблема часто не в последовательности гена, а в его эпигенетической регуляции. Это меняет всю логику терапевтического подхода.
Текущие направления исследований и лечения включают несколько стратегий:
- Реактивация молчащей копии гена. При синдроме Ангельмана материнская копия UBE3A заблокирована импринтингом, но физически присутствует в нейронах. Исследователи работают над тем, чтобы снять эту блокировку с помощью антисмысловых олигонуклеотидов (ASO) и CRISPR.
- Редактирование эпигенетических меток. Эпигенетическая пластичность рассматривается как перспективное направление: если метки можно установить, их теоретически можно и изменить. Инструменты на основе CRISPR-dCas9 позволяют точечно менять метилирование без разрезания ДНК.
- Симптоматическая терапия через метаболические цепочки. Текущие препараты для болезней импринтинга корректируют симптомы через метаболические пути, не изменяя ДНК напрямую. Это доказывает, что частичная терапия возможна даже без прямого редактирования генома.
- Технические ограничения генной терапии. Большие гены сложно доставить в клетки с помощью вирусных векторов. Это стимулирует разработку альтернативных подходов: редактирования генов прямо в клетках пациента и РНК-терапии.
- Индуцированные плюрипотентные стволовые клетки (iPSC). Создание клеточных моделей болезни из клеток конкретного пациента позволяет тестировать терапии до их применения в клинике. Hopeatrarelabs использует именно этот подход для ультраредких заболеваний.
Понять, почему нет одобренных лечений для многих редких болезней, помогает осознать масштаб задачи. Разработка терапии для болезней импринтинга требует одновременного прогресса в молекулярной биологии, клинической генетике и технологиях доставки препаратов.
Наиболее реалистичный горизонт для первых одобренных эпигенетических терапий при болезнях импринтинга составляет 5–10 лет. Клинические испытания ASO при синдроме Ангельмана уже идут, и первые результаты выглядят обнадёживающе.
Ключевые выводы
Геномное импринтирование — это эпигенетический механизм, при котором нарушение родительских меток на ДНК вызывает редкие болезни, не выявляемые стандартным секвенированием.
| Пункт | Подробности |
|---|---|
| Определение импринтинга | Около 100 генов человека активны только в зависимости от родительского происхождения аллеля. |
| Главные примеры болезней | Синдромы Прадера-Вилли и Ангельмана вызваны нарушением импринтинга одного участка хромосомы 15. |
| Диагностика требует двух методов | Анализ метилирования ДНК вместе с полногеномным секвенированием выявляет патологии, невидимые по отдельности. |
| Причина не в образе жизни | Болезни импринтинга возникают из-за эпигенетических сбоев в зародышевых клетках, а не из-за действий родителей. |
| Перспективы терапии | Реактивация молчащей копии гена через ASO и CRISPR-dCas9 — наиболее реалистичный путь к лечению. |
Почему импринтинг меняет разговор о редких болезнях
За годы работы с темой редких заболеваний я убедился в одном: большинство семей приходят к генетику с ощущением вины. Они ищут ошибку в своём образе жизни, в питании, в решениях, принятых до беременности. Понимание импринтинга разрушает этот нарратив полностью.
Болезни импринтинга возникают из-за эпигенетических сбоев в зародышевых клетках, которые не связаны с поведением родителей. Это не просто медицинский факт. Это психологическое освобождение для семей, которые годами несли груз необоснованной вины.
Второй момент, который меня поражает: импринтинг показывает, насколько узкой была наша концепция генетической болезни. Мы привыкли думать о мутациях в последовательности ДНК. Но болезнь может возникнуть без единого изменённого нуклеотида. Только из-за того, что метка стоит не там. Это переворачивает диагностическую логику.
Третье наблюдение касается терапии. Эпигенетические метки, в отличие от мутаций ДНК, теоретически обратимы. Это означает, что болезни импринтинга могут оказаться более поддающимися лечению, чем классические генетические заболевания. Это не повод для наивного оптимизма, но это реальное основание для научного интереса и финансирования исследований.
Расширение осведомлённости о геномном импринтировании среди врачей первичного звена сократило бы диагностические задержки на годы. Это не требует новых технологий. Только знания.
— John
Исследуйте редкие болезни глубже с Hopeatrarelabs
Если вы или ваш близкий столкнулись с редким генетическим заболеванием, понимание механизмов импринтинга — это только начало пути.
Hopeatrarelabs создаёт персонализированные модели болезней из клеток конкретного пациента с помощью технологий iPSC и CRISPR. Это позволяет тестировать тысячи одобренных FDA препаратов, антисмысловые олигонуклеотиды и варианты генной терапии параллельно, не дожидаясь, пока наука догонит конкретный диагноз. На платформе Hopeatrarelabs Knowledge Resource собраны научные материалы, инструменты и данные о редких болезнях с генетическим уклоном. Это ресурс для пациентов, семей, врачей и исследователей, которым нужна не общая информация, а конкретные ответы.
Часто задаваемые вопросы
Что такое геномное импринтирование простыми словами?
Геномное импринтирование — это механизм, при котором один из двух генов (от матери или от отца) намеренно заблокирован. Активен только один аллель, и это определяется химическими метками, установленными ещё до рождения.
Почему происходит импринтирование?
Импринтинг возник в ходе эволюции как результат конфликта между отцовскими и материнскими генами за контроль над ростом плода. Отцовские гены стимулируют рост, материнские его ограничивают, и импринтинг закрепил этот баланс на молекулярном уровне.
Какие болезни вызывает нарушение импринтинга?
Нарушения импринтинга вызывают синдромы Прадера-Вилли, Ангельмана, Беквита-Видемана и Сильвера-Рассела. Все эти состояния связаны с потерей активной родительской копии гена на конкретном участке хромосомы.
Можно ли вылечить болезни геномного импринтинга?
Одобренного лечения, устраняющего причину, пока нет для большинства таких болезней. Клинические испытания антисмысловых олигонуклеотидов при синдроме Ангельмана продолжаются, и эпигенетическая пластичность меток открывает реальные терапевтические перспективы.
Как диагностируют нарушения импринтинга?
Диагноз требует анализа метилирования ДНК в сочетании с полногеномным секвенированием. Стандартное секвенирование не выявляет эпигенетические сбои, поэтому специальный запрос на тест метилирования критически важен для точной диагностики.