
Missing a single immunogenicity flag during gene therapy screening can disqualify an otherwise eligible patient or, worse, trigger a severe adverse immune event mid-trial. For rare diseases, where patient pools are already vanishingly small, those errors are not recoverable. Gene therapy screening encompasses preclinical vector and candidate screening, immunogenicity assays, potency testing, and patient eligibility screening for clinical trials, making it one of the most technically demanding workflows in modern medicine. This guide walks through every phase of that workflow, from regulatory groundwork to troubleshooting edge cases, so your team can build a screening program that holds up under regulatory scrutiny and delivers real results for patients who have no other options.
Table of Contents
- Key prerequisites for gene therapy screening
- Preclinical screening: Immunogenicity and candidate selection
- Establishing gene therapy product potency
- Patient eligibility screening for clinical trials
- Troubleshooting, edge cases, and critical safety notes
- What most gene therapy screening guides miss
- Extend your impact with RareLabs resources
- Frequently asked questions
Key Takeaways
| Point | Details |
|---|---|
| Screening is multi-phased | Gene therapy screening relies on coordinated preclinical, potency, and patient-level evaluations. |
| Immunogenicity is a key barrier | Early and comprehensive immunogenicity screening can broaden clinical trial eligibility and reduce failures. |
| Patient selection uses strict criteria | Genetic, immunological, and functional status all inform patient eligibility for clinical trials. |
| FDA guidance is essential | Following the latest FDA guidelines ensures screening protocols meet regulatory standards and trial success. |
| Edge cases require strategies | Proactively addressing neutralizing antibodies and other outliers can preserve trial enrollment and safety. |
Key prerequisites for gene therapy screening
Robust gene therapy screening does not start in the lab. It starts with documentation, personnel qualification, and a clear regulatory strategy. Before a single assay runs, your program needs to be anchored to the right guidance documents and staffed by people who understand what those documents actually require in practice.
FDA guidances emphasize preclinical safety assessments including genome editing safety via NGS, potency assurance, CMC for INDs, and early-phase trial design considerations. The three most actionable documents are: the 2023 Potency Assurance guidance, the 2013 Preclinical Assessment guidance, and the 2015 Early-Phase Trials guidance. Together, they define what FDA expects at each stage and where sponsors most commonly fall short. Reviewing these before study design, not after your first IND submission, is essential.
Baseline requirements you need in place before screening begins:
- Validated assays with documented specificity, sensitivity, and reproducibility
- Qualified laboratory personnel with documented training records
- IRB-approved protocol and informed consent forms tailored to gene therapy risks
- Established standard operating procedures (SOPs) covering vector handling, sample processing, and data management
- An established chain of custody for patient-derived materials and biological specimens
- Review of genetic confirmation requirements specific to the target disease
Materials and tools you will need:
| Category | Specific items |
|---|---|
| Vectors | AAV serotypes, lentiviral constructs, or non-viral delivery systems |
| Cell lines | Disease-relevant primary cells or iPSC-derived models |
| Immunogenicity kits | ELISA kits for NAb detection, ELISpot plates, PBMC isolation reagents |
| Sequencing platforms | NGS platforms for off-target analysis and vector genome mapping |
| SOPs and data systems | Validated LIMS, audit trails, regulatory submission templates |
Pro Tip: Schedule a pre-IND meeting with FDA early in study design. This single step can prevent months of protocol revision by surfacing regulatory expectations before you are too far into your assay development to pivot easily.
Preclinical screening: Immunogenicity and candidate selection
With your foundational setup ready, the next step is to rigorously screen gene therapy candidates in a preclinical setting. Immunogenicity is the single greatest driver of patient exclusion and safety failures in gene therapy, so your preclinical screening strategy must be tiered and methodical.
Preclinical screening includes in silico, in vitro, and in vivo immunogenicity assessments such as MAPPs, PBMC stimulation, and ELISpot assays to predict immune responses to AAV capsids and transgenes. Each tier in the screening cascade serves a distinct purpose, and skipping any one of them significantly increases your risk of late-stage failures.
Comparison of preclinical immunogenicity methods:
| Method | What it detects | Sensitivity | Stage |
|---|---|---|---|
| In silico (bioinformatics) | Peptide binding predictions, MHC epitopes | Low to moderate | Earliest |
| MAPPs assay | MHC-II presented peptides from AAV capsids | High | In vitro |
| PBMC stimulation | T-cell proliferative responses | Moderate to high | In vitro |
| ELISpot | Antigen-specific cytokine-secreting T cells | Very high | In vitro or ex vivo |
| Murine model challenge | In vivo immune activation, toxicity | Variable by model | In vivo |
For comparison of viral and non-viral vectors, your immunogenicity risk profile changes substantially. AAV vectors carry well-characterized capsid immunogenicity challenges, particularly with pre-existing antibodies from natural exposure. Non-viral delivery options reduce some of those risks but introduce their own complexity around intracellular trafficking and expression efficiency.
Stepwise preclinical screening process:
- Select the vector based on tropism, payload size, and manufacturing feasibility
- Run in silico screens to flag high-risk peptide sequences in the capsid or transgene
- Perform MAPPs assay to identify peptides presented via MHC-II
- Conduct PBMC stimulation assays using donor blood that spans diverse HLA haplotypes
- Confirm T-cell response profiles with ELISpot
- Validate results in a relevant murine or non-human primate model
- Compare immunogenic profiles across candidate vectors and select the lead
Your immunogenicity assay approaches should also account for the transgene itself, not just the capsid. Expressed proteins can trigger immune activation independent of vector-related responses, particularly in diseases where patients have never expressed any functional version of the target protein.
Pro Tip: Reducing CpG content in your vector sequence is one of the most underused tactics in preclinical design. CpG-rich sequences activate TLR9 signaling, which drives innate immune responses that can confound immunogenicity results and reduce transgene durability. Many teams catch this in troubleshooting when it should be addressed at the design stage.
Establishing gene therapy product potency
Candidate vectors are now selected. The next phase is ensuring those products consistently perform through validated potency testing. Potency is not just a regulatory checkbox. It is your evidence that the product does what it claims to do, at the dose you plan to administer.
Potency assays for gene therapy products must reflect mechanism of action (MoA), evolve phase-appropriately from surrogate to validated functional assays, avoiding outdated methods like TCID50. Using TCID50 for AAV, which was designed for replicating viruses, is a mistake that still appears in early-stage programs and draws regulatory scrutiny almost immediately.
"Strategic cell line selection for permissivity and MoA relevance is the foundation of a defensible potency program. A potency assay run in a cell line that does not reflect your target tissue tells you almost nothing about what will happen in the patient."
What a strong potency strategy includes:
- A surrogate assay for early-phase submissions, such as transduction efficiency or genome copies per cell, that can be run quickly and reproducibly
- A functional readout assay tied directly to the therapeutic MoA, such as enzyme activity restoration, protein expression level, or a disease-relevant phenotypic endpoint
- Defined acceptance criteria that are pre-specified and justified before the assay runs
- A phase-appropriate validation plan, with full qualification at Phase 3 and beyond
Pitfalls to avoid:
- Using cell lines with no biological relevance to the target tissue
- Running potency assays on vector preparations that have not been properly titered
- Conflating infectious titer with biological potency
- Allowing potency assay formats to change between phases without bridging studies
- Failing to document lot-to-lot variability, which regulators will examine closely during BLA review
Poor potency data is one of the leading causes of clinical holds and IND rejections. The downstream cost of a clinical hold, in both time and resources, far outweighs the investment required to build a rigorous potency program at the start.
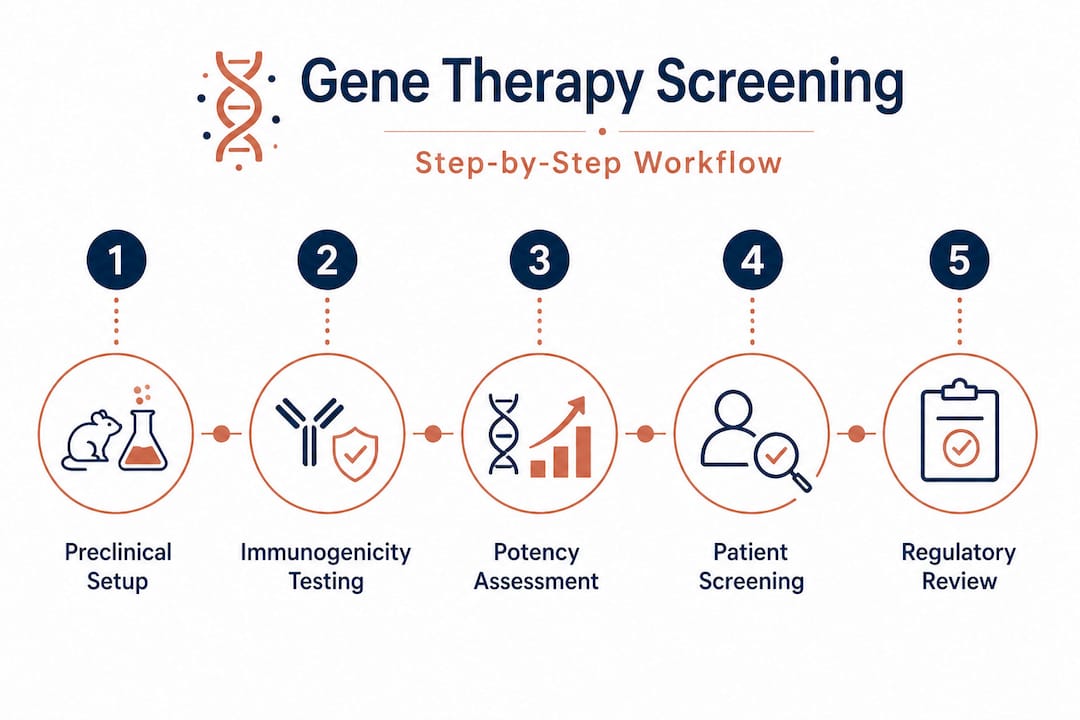
Patient eligibility screening for clinical trials
Ensuring your candidates are effective is essential. Just as crucial is making sure your patient selection process is equally robust. For rare diseases, where you might be working with a global eligible population of only a few hundred individuals, every eligibility criterion carries enormous weight.

Patient screening for eligibility in gene therapy trials for rare diseases involves genetic confirmation of mutation, absence of neutralizing antibodies (NAbs) to AAV, preserved organ function, no prior gene therapy, and suitable disease stage. Each of these parameters functions as a gate, and missing the evaluation of any one of them creates risk for the patient and liability for the program.
Step-by-step patient eligibility workflow:
- Collect full medical history, including prior biologics exposure and any history of AAV-related illness
- Confirm disease genotype through orthogonal genetic testing (both sequencing and deletion/duplication analysis)
- Perform NAb assay against the intended AAV serotype, using a validated ELISA with a pre-specified titer cutoff
- Assess organ function through standard clinical labs, targeting thresholds such as eGFR ≥60 for renal function or ALT/AST within defined limits for hepatic eligibility
- Apply disease staging criteria appropriate to the condition, such as mFARS score for ataxia trials or audiological thresholds for hearing loss programs
- Complete exclusion checks covering contraindicated medications, prior gene therapy, and active infections
Patient eligibility parameter table:
| Parameter | Typical threshold | Clinical rationale |
|---|---|---|
| Genetic confirmation | Biallelic pathogenic variants confirmed | Ensures diagnosis accuracy |
| AAV NAb titer | Below pre-specified cutoff (e.g., 1:50) | Prevents immune clearance of vector |
| Renal function (eGFR) | ≥60 mL/min/1.73m² | Supports vector clearance and safety monitoring |
| Liver enzymes | ≤2x ULN | Critical for hepatotropic vectors |
| Disease stage | Defined by validated rating scale | Ensures capacity to benefit |
| Prior gene therapy | None | Eliminates unpredictable immune priming |
Patient enrollment criteria for rare disease trials frequently require customization beyond standard oncology or common disease frameworks. The rarity of the disease means you cannot afford to set thresholds that unnecessarily exclude the only candidates available.
Pre-existing NAbs exclude patients (major eligibility limiter); high vector doses risk immune responses; CpG content in vectors triggers TLR9 immunity, reduced by depletion for longevity. These interconnected factors mean eligibility screening and preclinical design decisions must be coordinated, not treated as separate workflows.
Pro Tip: When NAb titers fall in a borderline range, some programs have successfully used plasmapheresis to transiently reduce circulating antibody levels before dosing. This is an edge case strategy, but it has enabled enrollment of patients who would otherwise be excluded, without meaningfully compromising safety when combined with careful monitoring.
Troubleshooting, edge cases, and critical safety notes
Even with robust protocols, tricky scenarios and pitfalls can emerge. Here is how to navigate and troubleshoot them effectively.
Mechanisms prioritize multi-tiered immunogenicity screening early to expand eligible populations and mitigate safety risks in AAV therapies for rare diseases. Overlooking NAb screening alone can exclude up to 30% of otherwise eligible rare disease patients, a proportion that is simply not recoverable when total eligible populations are already small.
Top troubleshooting checks for gene therapy screening programs:
- Verify NAb assay format is validated and serotype-matched to your vector
- Confirm your immunogenicity assays use PBMCs from a donor panel with diverse HLA representation
- Review organ function labs for timing; draw too early or too late relative to other medications and results may not reflect true baseline
- Check for discordance between genetic test reports and clinical presentation, which may indicate a second molecular diagnosis
- Audit your potency assay data for inter-run variability that exceeds acceptable limits
- Confirm that informed consent language accurately reflects the gene therapy risks specific to your vector and route of administration
Safety warning: Administering high-dose AAV in the presence of low-titer antibodies, even below your formal cutoff, can trigger acute immune complex deposition and complement activation. Mandatory post-dose monitoring at 24, 48, and 72 hours, including inflammatory markers and liver function tests, is not optional in high-dose systemic delivery programs.
80% hearing improvement rates observed post-screening in the OTOFERLIN (OTOF) gene therapy trial underscore what rigorous eligibility and immunogenicity screening can achieve when the program is designed right from the start. That outcome was not coincidental. It reflects careful patient selection, validated potency, and controlled vector immunogenicity working together.
What most gene therapy screening guides miss
Most gene therapy screening guides do an adequate job covering technical procedures. What they consistently fail to address is the strategic dimension that actually determines whether a program succeeds or stalls.
Patient selection balances disease severity with capacity to benefit, incorporating real-world data and early regulatory engagement. This is the "Goldilocks" cohort problem, and it is underappreciated. Include patients who are too severely affected and they may lack the residual cellular substrate to respond to gene therapy. Include only mildly affected patients and your efficacy readout will be underpowered because the therapeutic signal is too small relative to baseline function.
Most programs set eligibility criteria based on what is scientifically conservative or administratively convenient, not what maximizes the probability of demonstrating meaningful clinical benefit in the fewest possible patients. That is a strategic error. The right cohort is the one where the intervention has the best chance of producing a detectable, clinically meaningful response, and identifying that cohort requires real-world patient selection data, natural history data, and input from clinical experts who have actually managed the disease.
Early regulatory engagement is the other underemphasized lever. Teams that wait until after their Phase 1 protocol is locked to engage FDA often discover that their patient selection criteria, their potency assay format, or their immunogenicity monitoring plan does not align with current agency thinking. Mid-trial redesigns are expensive, disruptive to enrolled patients, and sometimes fatal to program timelines. A pre-IND meeting costs almost nothing by comparison. The programs that consistently move fastest are the ones that treat regulatory dialogue as a design input, not an obstacle to manage after the fact.
Extend your impact with RareLabs resources
Applying a rigorous gene therapy screening workflow is exactly the kind of work that accelerates real outcomes for rare disease patients who have no approved treatments and no time to wait for slow-moving pipelines.
RareLabs builds patient-specific disease models using iPSCs and CRISPR gene editing, then runs parallel treatment screens across FDA-approved drugs, custom ASOs, and gene therapy options to find viable strategies for ultra-rare conditions. If you are developing a screening protocol, evaluating trial eligibility criteria, or searching for evidence to support a clinical decision, the RareLabs knowledge database provides research-grade resources built specifically for rare disease programs. The RareLabs treatment search portal connects researchers and clinicians with active screens, trial tools, and actionable data to move faster and with greater precision.
Frequently asked questions
What is immunogenicity screening and why is it important in gene therapy?
Immunogenicity screening uses in vitro and in vivo assays such as MAPPs, PBMC stimulation, and ELISpot to predict how a patient's immune system will respond to the vector or transgene, preventing unexpected exclusions or dangerous immune reactions during the trial.
How are patients screened for eligibility in gene therapy trials?
Eligibility screening requires genetic confirmation, NAb testing, organ function labs, and disease staging, with each criterion validated against the specific vector, route of administration, and therapeutic target before enrollment begins.
Which FDA guidances are crucial for conducting gene therapy screening?
FDA guidances covering genome editing safety, potency assurance, CMC for INDs, and early-phase trial design are the core regulatory documents every gene therapy screening program must be built around from the earliest planning stages.
What are edge cases that complicate gene therapy trial enrollment?
Pre-existing NAbs, high vector doses, and CpG-driven TLR9 immune activation represent the most common edge cases that push otherwise eligible patients outside enrollment thresholds, requiring creative but safety-conscious protocol design to address.
How has gene therapy screening impacted trial outcomes?
Well-designed screening programs have produced outcomes such as 80% hearing improvement in OTOF gene therapy trials, demonstrating that precise patient selection and rigorous immunogenicity control translate directly into measurable clinical success.