
Un endpoint thérapeutique en maladie rare est un critère de mesure prédéfini, utilisé pour démontrer l'efficacité d'un traitement dans une population de patients souvent très réduite. La FDA et l'ANSM exigent que ces critères soient biologiquement justifiés, mesurables et adaptés aux contraintes spécifiques des essais cliniques en maladies orphelines. L'essai LEAP2MONO de Sanofi, conduit sur 43 patients atteints de la maladie de Gaucher type 3, illustre concrètement comment une architecture multidimensionnelle d'endpoints peut sécuriser des preuves d'efficacité là où les essais classiques échouent. Maîtriser ces critères d'évaluation des maladies rares est aujourd'hui une compétence centrale pour tout chercheur ou clinicien impliqué dans le développement thérapeutique.
Qu'est-ce qu'un endpoint thérapeutique en maladie rare ?
Un endpoint thérapeutique, ou critère de jugement, est la mesure principale sur laquelle repose la décision réglementaire d'approbation d'un traitement. Dans les maladies rares, ce critère doit être adapté à la maladie : les populations sont trop petites pour appliquer les standards statistiques habituels des essais à grande échelle.
La FDA rappelle que, depuis l'Orphan Drug Act de 1983, des centaines de traitements ont été approuvés pour des maladies rares. Pourtant, la majorité des maladies rares reste sans traitement validé. Ce constat souligne l'urgence de définir des endpoints pertinents, capables de prouver un bénéfice clinique même dans des cohortes de quelques dizaines de patients.

Le terme anglais endpoint est largement utilisé dans la littérature réglementaire francophone. Son équivalent direct est "critère de jugement principal" ou "objectif thérapeutique". Les deux formulations coexistent dans les dossiers soumis à l'ANSM et à l'Agence européenne des médicaments (EMA).
Quels types d'endpoints sont utilisés en maladies rares ?
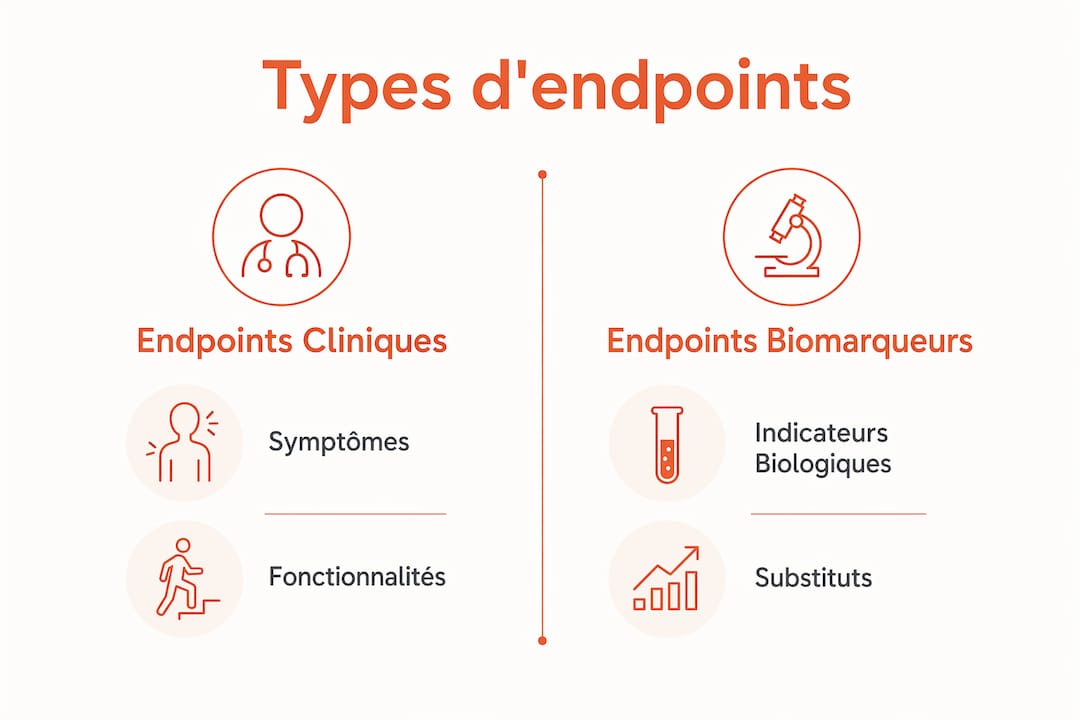
Les critères de jugement se divisent en deux grandes catégories : les endpoints primaires, qui déterminent le succès ou l'échec de l'essai, et les endpoints secondaires, qui documentent des effets complémentaires. Dans les maladies rares, cette architecture est souvent plus complexe qu'en médecine courante.
Les principaux types d'endpoints utilisés dans les essais cliniques en maladies orphelines sont :
- Biomarqueurs biologiques : mesures quantitatives d'un processus pathologique, comme les taux de GL1 et lyso-GL1 dans la maladie de Gaucher. Ils reflètent la pharmacodynamie du traitement.
- Mesures fonctionnelles : évaluations standardisées de la capacité motrice, neurologique ou respiratoire. Le score SARA modifié, utilisé dans LEAP2MONO, mesure l'ataxie cérébelleuse de façon reproductible.
- Critères cognitifs : outils comme le RBANS (Repeatable Battery for the Assessment of Neuropsychological Status), qui quantifient les fonctions cognitives dans les maladies à atteinte neurologique.
- Résultats rapportés par les patients (PRO) : mesures de qualité de vie ou de symptômes perçus, recueillis directement auprès des patients. Ces critères rapportés par les patients gagnent en importance réglementaire.
- Endpoints composites : combinaison de plusieurs mesures pour capturer un effet global, utile quand aucun critère unique n'est suffisamment sensible.
L'enjeu n'est pas de trouver un endpoint parfait. L'objectif est de sélectionner des critères validés biologiquement, capables de prouver un changement clinique significatif même dans de très petites cohortes.
Conseil de pro: Chaque endpoint doit être accompagné d'une justification biologique explicite dans le protocole. Les agences réglementaires évaluent la cohérence entre le mécanisme d'action du traitement et les critères retenus, pas seulement leur faisabilité technique.
Comment la FDA et l'ANSM encadrent-elles les endpoints en maladies rares ?
La FDA et l'ANSM ont toutes deux renforcé leurs dispositifs d'accompagnement en 2026, reconnaissant que les maladies rares nécessitent des cadres réglementaires spécifiques.
Du côté américain, le FDA Rare Disease Endpoint Advancement (RDEA) Pilot Program accepte des soumissions jusqu'au 30 juin 2026. Ce programme soutient la création d'endpoints novateurs pour les maladies rares, en favorisant ceux qui présentent un potentiel d'application large. Les endpoints retenus doivent pouvoir soutenir une décision réglementaire future, ce qui implique une validation méthodologique rigoureuse dès la conception.
En France, l'ANSM a lancé un dispositif fast-track national ciblant les essais de phase I et II en maladies graves et rares. Ce dispositif accélère l'évaluation des protocoles et influence directement le choix des endpoints, en imposant des conditions sur la nature des essais éligibles.
Les pratiques recommandées par les deux agences convergent sur plusieurs points :
- Réunions pré-IND et de type C : la FDA recommande une collaboration réglementaire précoce pour valider les endpoints, les plans d'évidence et les stratégies adaptatives avant le lancement de l'essai.
- Validation de la population cible : les agences exigent que la définition de la cohorte soit cohérente avec les endpoints retenus.
- Monitoring proportionné au risque : le cadre réglementaire 2026 insiste sur un suivi basé sur des critères prédéfinis, incluant biomarqueurs, mesures cliniques et données numériques.
Conseil de pro: Engagez le dialogue avec la FDA ou l'ANSM avant de finaliser votre stratégie d'endpoints. Une réunion de type C peut éviter des années de redéveloppement si un critère clé est jugé insuffisamment justifié.
Quels défis méthodologiques posent les essais cliniques en maladies orphelines ?
Les essais randomisés contrôlés classiques sont souvent impossibles dans les maladies ultra-rares. Les effectifs sont trop faibles pour atteindre la puissance statistique nécessaire. Cette contrainte fondamentale oblige à repenser l'architecture des preuves d'efficacité.
L'essai LEAP2MONO de Sanofi illustre cette approche. Sur 43 patients atteints de la maladie de Gaucher type 3, l'essai combine un score neurologique modifié (SARA) comme endpoint primaire, un score cognitif (RBANS) comme endpoint secondaire clé, et des biomarqueurs systémiques (GL1, lyso-GL1). Cette architecture multidimensionnelle permet d'atténuer l'incertitude sur l'effet principal et de documenter un changement cliniquement significatif à 52 semaines.
Architecture type d'un essai en maladie rare
| Niveau | Type d'endpoint | Exemple LEAP2MONO |
|---|---|---|
| Primaire | Fonctionnel/neurologique | Score SARA modifié |
| Secondaire clé | Cognitif | Score RBANS |
| Secondaire systémique | Biomarqueur | GL1, lyso-GL1 |
| Exploratoire | Qualité de vie | PRO spécifiques |
Les stratégies méthodologiques recommandées pour gérer ces contraintes sont les suivantes :
- Utiliser des contrôles externes : les données d'histoire naturelle de la maladie servent de référence comparative quand un groupe contrôle randomisé est impossible. Cette approche est validée par la FDA pour les essais en bras unique.
- Prédéfinir tous les critères : les seuils de succès, les fenêtres d'évaluation et les règles d'arrêt doivent être fixés avant le début de l'essai. Toute modification post-hoc fragilise la crédibilité réglementaire.
- Standardiser les mesures inter-sites : dans les essais multinationaux sur de petits effectifs, une variabilité de mesure même minime peut masquer un effet réel. La cadence des évaluations doit être identique sur tous les sites.
- Consolider avec des endpoints secondaires : les essais en petits effectifs utilisent un ensemble de secondaires pour renforcer les preuves quand l'effet primaire est modeste ou variable.
Les régulateurs exigent une preuve causale solide de l'endpoint, reposant sur standardisation, cadence des évaluations et contrôles externes rigoureux pour gérer l'incertitude par conception.
Conseil de pro: La standardisation inter-sites est le point de défaillance le plus fréquent dans les essais rares multinationaux. Investissez dans la formation des évaluateurs et dans des procédures opératoires standardisées dès la phase de setup.
Comment les thérapies innovantes redéfinissent-elles les objectifs thérapeutiques rares ?
La FDA a publié en 2026 un cadre pour accélérer le développement des thérapies ultra-rares individualisées. Ce cadre FDA 2026 cible explicitement l'édition génomique, les thérapies à base d'ARN et les oligonucléotides antisens (ASO), en soutenant l'évidence clinique quand les essais randomisés sont impossibles.
Ce cadre introduit des implications directes pour les endpoints :
- Endpoints de substitution (surrogate endpoints) : des biomarqueurs moléculaires ou génomiques peuvent être acceptés comme preuves d'efficacité si leur lien avec le bénéfice clinique est démontré.
- Endpoints novateurs : le RDEA Pilot Program redéfinit l'évidence exigée, en valorisant la justification et la validité des critères plutôt que leur seule mesurabilité.
- Données numériques et capteurs : les mesures collectées via dispositifs connectés entrent progressivement dans les dossiers réglementaires, notamment pour les maladies neuromusculaires.
- Approches adaptatives : les essais avec réévaluation intermédiaire des endpoints permettent d'ajuster la stratégie probatoire sans compromettre l'intégrité statistique.
L'impossibilité fréquente de conduire des essais randomisés dans les maladies ultra-rares pousse à construire une architecture de preuves solide et diversifiée, clé de crédibilité pour les approbations FDA et EMA. Les technologies comme les iPSC et l'édition CRISPR, utilisées par des acteurs comme Hopeatrarelabs, permettent de valider une cible thérapeutique en amont de l'essai clinique, renforçant la justification biologique des endpoints retenus.
Points clés
La maîtrise des endpoints thérapeutiques en maladies rares repose sur une architecture multidimensionnelle, une validation réglementaire précoce et une justification biologique rigoureuse de chaque critère retenu.
| Point | Détails |
|---|---|
| Définition des endpoints | Un critère de jugement doit être biologiquement justifié et adapté à la taille de la cohorte. |
| Architecture multidimensionnelle | Combiner endpoints primaires, secondaires et biomarqueurs renforce la crédibilité des preuves. |
| Engagement réglementaire précoce | Les réunions pré-IND avec la FDA ou l'ANSM réduisent le risque de rejet des critères retenus. |
| Cadres 2026 FDA et ANSM | Le RDEA Pilot Program et le fast-track ANSM accélèrent la validation des endpoints novateurs. |
| Standardisation inter-sites | Une cadence et des procédures uniformes sur tous les sites sont indispensables dans les petits effectifs. |
Ce que l'expérience m'a appris sur les endpoints en maladies rares
Après des années à suivre le développement de programmes thérapeutiques en maladies rares, un constat s'impose : la plupart des échecs réglementaires ne viennent pas d'un traitement inefficace. Ils viennent d'endpoints mal choisis ou insuffisamment justifiés.
La tentation est forte de sélectionner des critères faciles à mesurer plutôt que des critères cliniquement pertinents. Un biomarqueur sanguin est plus simple à collecter qu'un score neurologique standardisé. Mais si ce biomarqueur ne reflète pas un bénéfice perçu par le patient, les agences le rejetteront, et à juste titre.
Ce que j'observe également, c'est que les équipes qui réussissent engagent le dialogue réglementaire très tôt, parfois avant même de finaliser le protocole. Une réunion de type C avec la FDA n'est pas une formalité administrative. C'est un investissement qui peut remodeler toute la stratégie d'évidence et éviter des années de développement inutile.
L'autre angle mort fréquent concerne les résultats rapportés par les patients. Ces critères sont souvent traités comme des endpoints exploratoires de second rang. Or, dans les maladies rares où l'impact fonctionnel est central, un PRO bien construit peut devenir l'argument le plus convaincant du dossier réglementaire. Les agences y sont de plus en plus attentives.
La prochaine évolution majeure sera l'intégration systématique des données numériques comme endpoints de surveillance continue. Les capteurs portables et les outils de mesure à distance vont transformer la façon dont on documente l'efficacité dans les essais à petits effectifs. Les équipes qui s'y préparent maintenant auront une longueur d'avance décisive.
— John
Hopeatrarelabs : ressources pour vos programmes en maladies rares
Les professionnels qui développent des thérapies en maladies rares ont besoin d'un accès rapide aux mises à jour réglementaires et aux méthodologies d'endpoints validées. Hopeatrarelabs met à disposition une base de ressources spécialisées pour accompagner cette démarche.
La RareLabs Knowledge Resource centralise les informations réglementaires 2026, les approches méthodologiques et les données sur les thérapies en développement pour les maladies ultra-rares. Hopeatrarelabs développe également des modèles de maladie personnalisés à partir de cellules souches pluripotentes induites (iPSC) et de l'édition CRISPR, permettant de tester des milliers de molécules en parallèle et de renforcer la justification biologique des endpoints avant l'entrée en essai clinique. Pour les équipes qui cherchent à accélérer leur programme, la plateforme Hopeatrarelabs offre une expertise directement applicable à la conception des critères de jugement.
Questions fréquentes
Qu'est-ce qu'un endpoint thérapeutique en maladie rare ?
Un endpoint thérapeutique est un critère de mesure prédéfini utilisé pour évaluer l'efficacité d'un traitement dans un essai clinique. En maladie rare, ce critère doit être biologiquement justifié et adapté aux petites cohortes.
Quelle est la différence entre endpoint primaire et secondaire ?
L'endpoint primaire détermine le succès ou l'échec de l'essai. Les endpoints secondaires documentent des effets complémentaires et renforcent les preuves d'efficacité quand l'effet primaire est modeste.
Comment la FDA valide-t-elle les endpoints en maladies rares ?
La FDA recommande des réunions précoces de type pré-IND ou Type C pour valider les endpoints avant le lancement de l'essai. Le RDEA Pilot Program 2026 soutient spécifiquement le développement d'endpoints novateurs pour les maladies rares.
Peut-on utiliser des biomarqueurs comme endpoints principaux ?
Oui, sous conditions. Un biomarqueur peut être accepté comme endpoint de substitution si son lien avec un bénéfice clinique significatif est démontré. Le cadre FDA 2026 pour les thérapies ultra-rares individualisées élargit cette possibilité pour les thérapies génomiques et ARN.
Qu'est-ce qu'un essai en bras unique et quand est-il justifié ?
Un essai en bras unique est un essai sans groupe contrôle randomisé, justifié quand la maladie est trop rare pour constituer deux groupes. Il repose sur des données d'histoire naturelle comme référence comparative et exige une justification réglementaire explicite.